
法米替尼治疗晚期实体瘤患者的药代动力学及安全性
2024-01-10 06:19唐银梅冯元莹白桂颖王璇赵伟鹏张杰史业辉
天津医科大学学报 2024年1期
唐银梅,冯元莹,白桂颖,王璇,赵伟鹏,张杰,史业辉
(天津医科大学肿瘤医院乳腺内科,国家恶性肿瘤临床医学研究中心,天津市肿瘤防治重点实验室,天津市恶性肿瘤临床医学研究中心,乳腺癌防治教育重点实验室,天津 300060)
受体酪氨酸激酶(receptor tyrosine kinase,RTK)是一个跨膜蛋白家族,在许多细胞生物过程中至关重要,包括增殖、分化、迁移、血管生成和细胞存活等[1]。许多肿瘤的发生、发展都与酪氨酸激酶的异常表达密切相关。舒尼替尼是一种多靶点酪氨酸激酶抑制剂,具有强大的抗血管生成作用和抗肿瘤活性,已于2006 年被美国食品药品监督管理局(FDA)批准用于肾细胞癌和胃肠道间质瘤[2]的治疗。临床研究表明,单独应用舒尼替尼对多种恶性实体肿瘤包括肾癌、对伊马替尼耐药的胃肠道间质瘤和晚期胰腺神经内分泌肿瘤等有效[2-4]。但舒尼替尼的严重或危及生命的不良事件已被报道,甚至FDA 也在舒尼替尼中增加了一个关于肝毒性的“黑匣子”警告的标签[5]。法米替尼是一种新型的多靶点酪氨酸激酶抑制剂,对多种RTK 表现出优异的抑制活性,包括干细胞因子受体、血管内皮生长因子受体(VEGFR)等。动物研究还表明,在人类肿瘤异种移植模型中,与单独使用5-氟尿嘧啶等化疗药物相比,法米替尼在抑制肿瘤生长方面的效力更高[6]。由于Ⅰ期临床剂量所采用的是不同小规格的剂量组合(1、4、12 mg),且剂量并未涵盖Ⅲ期注册研究所有剂量。因此本研究以同位素内标为参照,开展法米替尼15、20、25mg剂量组在晚期实体瘤患者的药代动力学特征和安全性,旨在完善药代特征描述并为临床应用提供依据。
1 对象与方法
1.1 研究对象 本研究中,46 例受试者签署了知情同意书进行筛选,10 例受试者不符合入选/排除标准,4 例受试者撤回了知情同意,最后32 例受试者经筛选合格进行了入组。纳入标准:(1)年龄≥18岁且≤70 岁,组织学确诊并且经标准治疗无效或目前无标准治疗手段的晚期或转移的实体瘤患者。(2)ECOG 体力状况评分:0~1 分。(3)预计生存期≥3 个月。(4)主要器官功能良好,首次用药7 d 内相关指标符合以下标准:①中性粒细胞计数≥1 500/mm3或≥1.5×109/L。②血小板计数≥100 000/mm3或≥100×109/L。③血红蛋白≥90 g/L(14 d 内未输血)。④血清总胆红素≤1.5×正常值上限(ULN)。⑤血清肌酐≤1.5×ULN。⑥血谷丙转氨酶(ALT)和血谷草转氨酶(AST)分别≤2×ULN;如存在肝转移,则为≤5×ULN。排除标准:(1)签署知情同意书前3 周内接受过同类VEGFR 类小分子酪氨酸激酶抑制剂(如法米替尼、索拉非尼、舒尼替尼、瑞戈非尼等)治疗的患者。(2)患有高血压且经两种降压药物治疗无法获得良好控制者(收缩压>140 mmHg,舒张压>90 mmHg,1 mmHg=0.133 kPa)。(3)已知或怀疑对法米替尼或同类药物过敏。该研究方案已获得天津医科大学肿瘤医院医学伦理委员会批准(E2016151)。所有患者都对该研究提供了书面知情同意书。
1.2 研究方法 本研究为一项开放性临床试验。代谢产物SHR116637 有药效活性,评估法米替尼15、20 与25 mg/d 及代谢物SHR116637 单次与多次给药的药代动力学特征与药代动力学参数。单次给药主要参数:(1)药物浓度-时间曲线。(2)消除半衰期(t1/2)。(3)体内平均滞留时间(MRT)。(4)表观分布容积(Vd/F)。(5)全身清除率(CL/F)。(6)达峰浓度(Cmax)、达峰时间(Tmax)。多次给药主要参数:(1)药物浓度-时间曲线。(2)Tmax。(3)稳态谷浓度(Css,min)。(4)稳态峰浓度(Css,max)。(5)消除半衰期(t1/2)。(6)稳态血浆浓度-时间曲线下面积(AUCss)。(7)全身清除率(CL/F)。记录不良事件及严重不良事件的发生率。
1.2.1 法米替尼给药方案 法米替尼用法和用量:空腹,口服给药,每日1 次。单次给药:受试者于给药前1 日晚起开始禁食10 h,不禁水过夜,第1 天(单次给药当天)早晨采集空白血样后口服法米替尼,用240 mL 温水送服。按照前期试验结果[7],法米替尼的半衰期为33 h,5 个半衰期需168 h 以上,故单次给药后洗脱至少7 d 进行多次给药。多次给药:每日1 次,连续给药28 d 为1 个周期。如果受试者能耐受且从中获益可以继续接受免费治疗,直至病情进展、不可耐受、受试者主动要求退出或研究者判断受试者不再适合接受研究药物治疗。
1.2.2 药代动力学(pharmacokinetics,PK)采血与浓度测定 为了确定法米替尼单次给药和多次给药阶段的PK 特征,每例受试者进行了PK 采血。单次给药阶段:第1 天给药前15 min 内和给药后1、2、3、4、5、6、8、12、24、48、72、96、120、144 h 采集血样(3 mL);连续给药阶段:在第13 天给药前15 min、第14 天给药前15min 内以及给药后1、2、3、4、5、6、8、12 和24 h收集血液样品(3 mL)。样本采集至肝素抗凝管中,4℃,3 500 r/min 离心10 min,取血浆分装,于-80℃低温冰箱保存。采用经过验证的高效液相色谱-质谱联用检测法(LC-MS/MS)检测血浆样品中法米替尼及其主要代谢物M3(SHR116637)的血药浓度[9]。
1.2.3 不良事件判定 按“肯定有关、可能有关、可能无关、肯定无关、无法判定”5 级分类法对不良事件和试验用药之间可能存在的关联做出评估。本研究中定义“肯定有关”、“可能有关”和“无法判定”均列为研究药物相关不良事件。不良事件根据药物临床试验质量管理规范进行判定。
1.3 统计学处理 本研究的主要分析为描述性统计汇总,不涉及正式的假设检验。使用WinNonlin 7.0 软件计算个体PK 参数并分剂量组对单、多次给药后血浆中法米替尼及其代谢物SHR-116637 的主要PK 参数进行描述性统计汇总。按照剂量组列表本次试验所发生的不良事件,对受试者例数及百分数进行描述性统计。
2 结果
2.1 入组患者人口统计学和基线特征 患者人口统计学和基线特征如表1 所示。受试者大部分为女性(30 例,93.8%),中位年龄54(33~65)岁,主要为乳腺癌(84.4%)。疾病临床分期均为晚期(Ⅲ期3例,Ⅳ期29 例),所有受试者均接受过至少1 次化疗治疗,68.8%的受试者接受过既往放疗,87.5%的受试者接受过手术。
2.2 单次和多次给药后血浆中法米替尼和代谢物SHR116637 的PK 特征
2.2.1 单次给药后血浆中法米替尼和代谢物SHR-116637 的PK 特征 单次口服法米替尼后,血浆中原型药物和代谢物SHR116637 的Tmax在6 h 内达峰。原型药体内分布广泛且消除慢。低、中、高剂量组单次给药后代谢物SHR116637 总体暴露量(以AUC0-t计)分别相当于原型药物法米替尼的9.91%、9.65%和8.30%,见图1、表2。

图1 单次给药阶段法米替尼及代谢物SHR116637 平均血药浓度-时间曲线Fig.1 Average plasma concentration-time curve of famitinib and metabolite SHR116637 in single administration stage
表2 单次给药后血浆中法米替尼和代谢物SHR116637 的PK 参数[M(P25,P75),]Tab.2 PK parameters of famitinib and metabolite SHR116637 in plasma after a single dose[M(P25,P75),]

表2 单次给药后血浆中法米替尼和代谢物SHR116637 的PK 参数[M(P25,P75),]Tab.2 PK parameters of famitinib and metabolite SHR116637 in plasma after a single dose[M(P25,P75),]
注:Cmax:达峰浓度;Tmax:达峰时间;AUC:药时曲线下面积;t1/2:消除半衰期;MRT:体内平均滞留时间;CL/F:全身清除率;Vd/F:表观分布容积
?
2.2.2 多次给药后血浆中法米替尼和代谢物SHR-116637 的PK 特征 低、中、高3 个剂量组多次给药后,血浆中法米替尼和代谢物SHR116637 的Tmax与单次给药相比略有延迟,Cmax、稳态暴露量(AUCss)随给药剂量增加而逐渐升高;多次给药后法米替尼和代谢物SHR116637 在体内存在一定程度的蓄积,以代谢物SHR116637 蓄积程度更明显,见图2、表3。
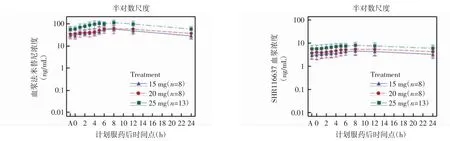
图2 多次给药法米替尼及代谢物SHR116637 平均血药浓度-时间曲线Fig.2 Average plasma concentration-time curve of famitinib and metabolite SHR116637 in multiple dosing stages
表3 多次给药后血浆中法米替尼PK 参数[,M(P25,P75)]Tab.3 PK parameters of famitinib and metabolite SHR116637 in plasma after multiple dosing[,M(P25,P75)]
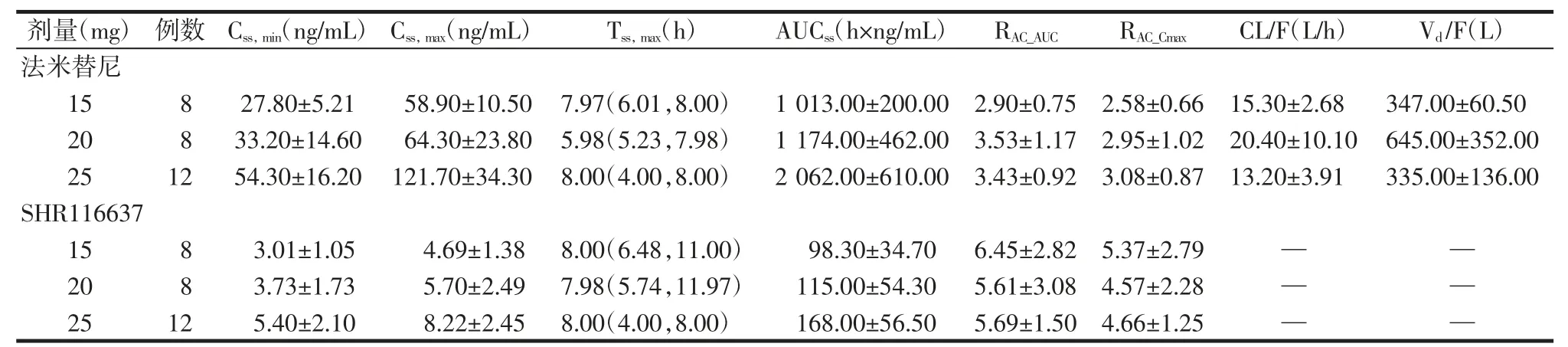
表3 多次给药后血浆中法米替尼PK 参数[,M(P25,P75)]Tab.3 PK parameters of famitinib and metabolite SHR116637 in plasma after multiple dosing[,M(P25,P75)]
注:Cmax:达峰浓度;Tmax:达峰时间;AUC:药时曲线下面积;CL/F:全身清除率;Vd/F:表观分布容积;RAC:药物蓄积比
?
2.3 不良事件
2.3.1 单次给药不良事件 单次给药组中,共发生5 例次3 级及以上药物相关的不良事件,分别为:血压升高、γ-谷氨酰转移酶升高、血红蛋白降低,其中血压升高最为常见,共发生3 例次,15 mg 剂量组2例次,20 mg 剂量组1 例次。单次给药阶段未出现因不良事件导致停药或退组的受试者。
2.3.2 多次给药不良事件 多次给药组中,共发生24 例次3 级及以上药物相关的不良事件,血压升高最常见,其次为中性粒细胞减少、血小板降低、白细胞降低等。2 例受试者因呼吸衰竭(1 例,20 mg)和心力衰竭(1 例,25 mg)死亡,考虑与研究药物相关。
3 讨论
Xie 等[9]研究了法米替尼在实体瘤患者体内的代谢及生物转化机制,发现法米替尼在人体内吸收缓慢,单次口服20 mg 法米替尼后原型药和主要代谢产物M3(SHR116637)达峰时间为(6.75±2.38)h和(7.75±2.71)h,Cmax分别为(21.7±7.78)、(0.96±0.36)ng/mL。本研究中单次给药剂量20 mg 法米替尼后原型药和代谢物SHR116637 均在5.98 h 内达峰,Cmax分别为(22.40±5.69)、(1.47±0.75)ng/mL。多次给药后法米替尼与代谢物SHR116637 均存在一定程度的蓄积,且以代谢物SHR116637 中15 mg 剂量组最为明显,蓄积指数RAC_AUC和RAC_Cmax分别为6.45±2.82 和5.37±2.39,该蓄积可能与较短的给药时间间隔(24 h)和较长的消除半衰期有关。对于多次给药,本研究考察的是连续服药14 d 的PK,Zhou等[7]研究考察的是连续服药28 d 的PK,在同等剂量下(25 mg),两个研究的原型药物法米替尼在受试者体内的稳态达峰时间基本一致,就稳态下暴露量(Cmax和AUC)而言,本研究中多次给药25 mg 法米替尼后,原型药的Cmax为(121.70±34.30)ng/mL,AUCss为(2 062.00±610.00)h×ng/mL 结果略高于Zhou 等研究结果[多次给药剂量25 mg,原型药Cmax为(70.5±26.60)ng/mL,AUC0-24为(1 173±486)h×ng/mL]。考虑到本试验的样本量有限,受试者疾病状态不同以及个体差异等,将在后续临床研究中进一步收集更多数据,通过群体药代动力学分析,考察可能影响法米替尼PK 的内外在因素,如人口统计学、疾病状态、合并用药、肝肾功能等。
本研究共设置15、20、25 mg 3 个剂量组。各剂量组单次给药及多次给药后法米替尼的安全性和耐受性在可接受范围内,并且3 个剂量组的不良事件发生率没有显著的剂量-效应关系。本研究中单次和多次口服法米替尼后,血压升高是与药物相关最常见的不良事件之一,发生率分别为9.4%、40.6%,这与前期Ⅰ期[7-8]耐受性研究中观察到药物相关的不良事件相吻合。据报道多激酶抑制剂(如舒尼替尼和索拉非尼)在治疗肿瘤过程中出现血压升高,与其对VEGFRs 的抑制作用有关[10]。临床中,在有效控制血压的同时要慎用硝苯地平和氨氯地平等钙离子拮抗剂,一方面该类药物对CYP3A4 代谢酶有弱抑制性,另一方面CYP3A4 主要参与法米替尼代谢清除,这可能会引起法米替尼暴露量的增加。其次,中性粒细胞减少(15.6%)在多次给药后也很常见,但是与常规化疗药物引起的中性粒细胞减少相比,下降程度较轻,对患者影响较小。Donskov等[11]发现在舒尼替尼治疗期间出现中性粒细胞减少和高血压,或在较小程度上发生手足综合征,预示着结局改善。在本研究中,单次和多次给药阶段也发生ST-T 段改变、疲劳、腹泻、手足综合征等不良反应,但是症状较轻、患者可耐受且停药后很快恢复。
本研究结果表明法米替尼的耐受性良好,相关不良反应是可预测和可控的,为法米替尼在临床上的合理应用奠定基础。但由于纳入的病例较少,且以乳腺癌为主,法米替尼在其他肿瘤中的应用尚需更多的临床验证。
猜你喜欢
中国心血管杂志(2022年2期)2022-11-25
中国心血管杂志(2022年4期)2022-11-25
现代临床医学(2022年4期)2022-09-29
中国经济周刊(2021年10期)2021-06-06
中国心血管杂志(2021年6期)2021-01-02
中国人口·资源与环境(2020年10期)2020-12-23
人物画报(2019年4期)2019-10-26
中国医药指南(2019年25期)2019-10-22
中国心血管杂志(2019年3期)2019-01-04
分析测试学报(2015年7期)2016-01-13
