
ZrMnFe 基吸气合金吸附二氧化碳气体的性能和机理
2024-01-23 04:32杨乔彬曾凡浩黄睿高亚芳
粉末冶金材料科学与工程 2023年6期
杨乔彬,曾凡浩,黄睿,高亚芳
(中南大学 粉末冶金研究院,长沙 410083)
能源和环境是困扰现代社会发展的两大问题[1-3]。人类对化石燃料的依赖日益增加,而化石燃料的消耗会产生大量的二氧化碳(CO2),是公认的主要温室气体之一,当下CO2吸收成为一个非常重要和热门的话题。目前使用较多的CO2吸附材料主要有活性炭、金属有机骨架材料(metal-organic frameuorks, MOFs)、聚合物、碳纳米管、介孔二氧化硅、胺基负载材料和金属间化合物等[4-8]。种类繁多的吸气材料中,AB2Laves 型金属间化合物(A为Ti、Zr、稀土金属、Mg,B 为 Fe、Co、Ni、Mn等其他过渡金属和非过渡元素)具有比表面积大、活性位点丰富、选择性优良和相对较高的质量储存容量等特点,受到众多科学研究人员的关注[9-10]。并且Laves 型金属间化合物含有360 多种二元化合物和540 多种三元化合物[11-12],通过使用不同元素替代A、B,可以得到三元甚至四元化合物,丰富了Laves 型金属间化合物对各种气体(如CO、CO2、O2、N2、CH4和H2O 等)吸附的选择性。BAKER 等[13]使用 Laves 型 ZrMnFe 基三元合金吸气剂ST909(SAES 公司,意大利)对含CO2的混合气体进行吸附,吸附率达到99.9 %。在AB2Laves 合金中,用Ti 部分取代Zr 被证明是改善Zr 基Laves 相合金氢化率的有效方法[14]。ZHANG 等[15]制备了Zr0.9-xTi0.4+xV1.7非化学计量储氢合金,发现Ti 部分取代Zr 改变了ZrV2的相组成和微观结构,提高了其吸氢能力。
CO2吸气剂表面的吸附和脱附过程非常复杂,且吸附反应很难通过实验观察,采用理论计算进行模拟和预测,可为实验提供有用的信息。LIU 等[16]用密度泛函理论(density functional theory, DFT)和范德瓦尔斯修正(DFT+VDW)计算方法研究CO2在TiO2表面上的吸附和还原,包括从单一吸附到在不同表面位置迁移的场景,结果表明还原的TiO2表面更容易吸附CO2。WU 等[17]基于DFT 的第一性原理计算,研究了CO2在Cu2O (111)表面氧空位的吸附,结果表明CO2不易吸附在氧空位,但CO2分子会在氧空位裂解形成自由基阴离子CO2δ-,从而更易吸附在其他位点。LI 等[18]成功合成了分别裸露于(110)和(001)表面的两种多孔ZnO,进行CO2的吸附对比实验,证明CO2的吸附对晶体的晶面有选择性,并使用第一性原理计算对其进行了选择性研究。QI等[19]在密度泛函理论框架内研究 CO2分子在δ-Pu (100)表面的吸附行为,发现δ-Pu (100)表面的最佳吸附位点为空位,吸附能为-6.430 eV,属于强化学吸附,并且CO2分子主要与Pu 表面的分子相互作用,与其他3 个Pu 原子的相互作用较弱。
基于ZrMnFe 基合金对低浓度的CO2有99.9 %的去除率,但对于纯CO2的吸附还缺乏充分的研究。本实验制备ZrMnFe 基合金并对其吸附CO2的行为进行研究,同时对比Ti 掺杂后吸附性能的变化,结合第一性原理对其吸附机理进行研究,为Laves 型金属间化合物与含CO2气体的反应提供理论和技术指导。
1 实验和计算
1.1 ZrMnFe 基合金粉末的制备
主要材料包括金属锆(Zr,国核宝钛锆业股份公司,管状)、金属钛(Ti,国核宝钛锆业股份公司,圆片)、金属铁(Fe,宜昌佳晟鑫铁合金有限公司,棒状)、电解锰(Mn,宜昌佳晟鑫铁合金有限公司,棒状)等高纯金属,纯度(质量分数)均大于99.9%,和CO2(湖南凯美特气体股份有限公司,体积分数>99.9%)。
按TixZr1-xMnFe(x=0、0.25,摩尔分数。下同)化学式比例计算出原料的质量比,用电子天平(FA2104,上海舜宇恒平科学仪器有限公司)准确称量金属原料共计约500 g。超声清洗后烘干,用真空非自耗电弧熔炼炉(DHL-400,中国科学院沈阳科学仪器股份有限公司)进行熔炼。熔炼前将炉内压力降低到 5×10-3Pa,然后充入氩气,直到压力增至0.05 MPa。熔炼电流设置为350 A。为保证样品成分均匀,熔炼过程中将铸锭翻转并重复熔化5 次以上。由于锰具有较高的蒸汽压,在熔化过程中会挥发损耗,因此熔炼过程中需取出半成品称重,计算锰的损耗量并加以补充,确保获得成分比相对准确的合金样品。为了改善枝晶偏析,熔炼后的合金铸锭在1 000~1 200 ℃均匀化热处理 16~24 h,随后通过振动破碎机(GJ-I,长沙天创粉末技术有限公司)制成粉末,将合金粉末过 200 目筛(筛网孔径为75μm)后,用压片机将粉末压制成直径为1.2 cm、厚度为0.1 cm 的片状样品。
1.2 组织与性能表征
1.2.1 CO2吸附实验
用自制的简易 Sieverts 型 PCT 测试仪对TixZr1-xMnFe 合金进行CO2吸附实验,当容器内压力稳定不变时停止实验,实验过程如图1 所示。反应器的总容积为533 mL,容器内无CO2气体时的初始压强约0.43 MPa,吸附过程中,反应器内的气压随着CO2吸附量增加而减小。通过PCT 测试仪的压力感应器实时监测压强,每4 h 记录一次,根据理想气体的状态方程(见式(1))计算容器内CO2气体的量,进而得到TixZr1-xMnFe 对CO2气体的吸附量,由此得到恒定温度下的CO2吸收动力学曲线。曲线中CO2的吸附量为单位质量合金吸收的CO2气体的量,即n(CO2)/m(TixZr1-xMnFe)。
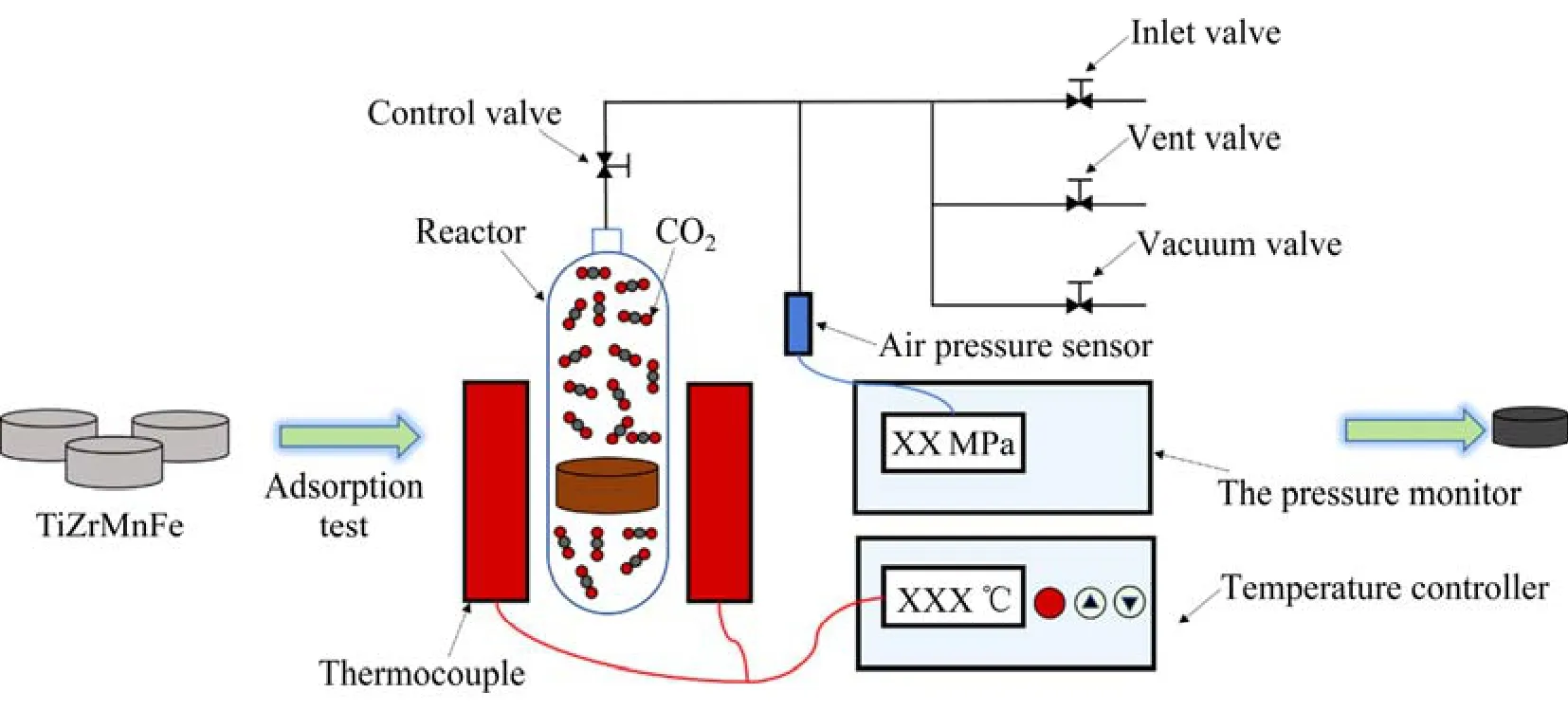
图1 CO2 吸附实验过程示意图Fig.1 Schematic diagram of CO2 absorption experiment
式中:P为反应器内的压强,Pa;V为气体体积553×10-4m3;n为气体的物质的量,mmol;T为反应器内的温度,K;R为摩尔气体常数,为8.314 J/(mol·K)。
通过温度控制器设置吸附环境温度,得到不同的吸附曲线,同时通过公式(2)计算吸附前后样品的质量增加率r,以此探究吸附反应发生的最佳温度。
式中:m1表示样品吸附前的质量,m2表示样品吸附后的质量。
1.2.2 X 射线衍射分析
采用XRD (Rigaku-3014,深圳市科时达电子科技有限公司)对吸气前后合金粉末的物相组成进行分析,取过 200 目孔筛后的样品粉末进行测试,XRD 扫描角度为 10°~90°,扫描速度为 5 (°)/ min。
1.2.3 扫描电子显微镜分析
将合金块体镶样预磨抛光处理,采用场发射扫描电子显微镜(Nova Nano SEM 230,上海禹重实业有限公司)表征热处理前后合金的微观组织结构,并通过能谱仪对元素组成与分布进行进一步分析。
1.2.4 气相色谱质谱分析
使用气体采样袋收集反应结束后的尾气,采用GCMS 型气相色谱质谱联用仪进行气相色谱质谱分析,与未反应的气体比对。利用总离子流色谱法(total ionization chromatography, TIC)记录具有某质荷比的离子强度随时间变化的图谱。在选定的质量范围内,任何一个质量数都有与总离子流色谱图相似的质量色谱图。由总离子色谱图可以得到任一组分的质谱图,由质谱图利用计算机在数据库中检索,可以得出几种最可能的化合物,包括化合物名称、分子式、分子量、基峰以及匹配度。
1.3 计算方法与模型
本文采用基于第一性原理的赝势平面波方法对TixZr1-xMnFe (110)表面吸附CO2分子进行模拟计算[20-22]。首先采用LBFGS (large-Broyden-Flecher-Goldfarb-Shanno)[23-25]共轭梯度方法的密度混合方案对构建体系的几何结构进行优化,得到稳定结构后再计算各吸附位置的能量,选取优化后的体系分别计算吸附能、电子态密度和差分电荷密度。体系的价电子波函数用平面波基矢展开并设平面波的截断能为500 eV,迭代收敛精度为2×10-6eV,选择广义梯度近似下的PBE (Perdew-Burke-Emzerhof)泛函来描述交换关联能[26-28],采用超软赝势计算离子实与电子之间的相互作用,总能量计算在倒易空间中进行,布里渊区积分采用Monkhorst-Pack方法,K 点取3×3×1[29]。文中的几何优化、吸附能计算和电子结构计算由CASTEP 量子模块完成。
ZrMn2是Laves 相除杂合金的代表之一,空间群为P6/mmc,晶胞参数为a=0.504 5 nm,b=0.485 6 nm,c=0.802 9 nm,α=β=90°,γ=120°[30]。TixZr1-xMnFe与ZrMn2具有相同的相结构,在晶胞模型构建中用Fe 取代部分Mn 得到ZrMnFe,再通过Ti 取代部分Zr 得到Ti0.25Zr0.75MnFe,过程如图2(a)所示。几何优化结构后得到 Ti0.25Zr0.75MnFe 的晶胞参数为a=0.494 3 nm,b=0.472 0 nm,c=0.825 1 nm。在此基础上对TixZr1-xMnFe 进行切取,得到其(110)表面结构。TixZr1-xMnFe (110)清洁理想表面结构如图2(b)所示,计算中采用6 个原子层的表面结构,设置真空层高度为1.5 nm,固定最下面4 层的坐标,其余原子允许弛豫。
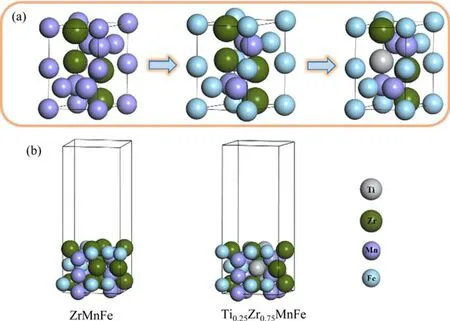
图2 TixZr1-xMnFe 晶胞模型构建(a)及(110)清洁表面模型(b)Fig.2 TixZr1-xMnFe cell model construction (a) and (110) clean surface model (b)
2 实验结果
2.1 TixZr1-xMnFe 合金的CO2 气体吸附性能
图3 所示为制备的TixZr1-xMnFe 合金的XRD图。对比JCPDS(PDF#65-2001) 中的 ZrMn2标准衍射峰,发现TixZr1-xMnFe 合金的各个衍射峰特征与ZrMn2的一致,这表明其相组成和晶体类型与ZrMn2一致。

图3 TixZr1-xMnFe 粉末的XRD 图谱Fig.3 XRD patterns of TixZr1-xMnFe powders
为了研究Ti掺杂对TixZr1-xMnFe 晶体结构的影响,根据布拉格定律计算其各个晶面的晶面间距:
式中:d为晶面间距,λ为入射波波长,θ为入射光和晶面之间的夹角。结果列于表1,可以发现Ti 掺杂后,各晶面的XRD 衍射峰均向高角度位置偏移,晶面间距也随之变短。这是由于Ti 原子半径小于Zr 的原子半径所致。
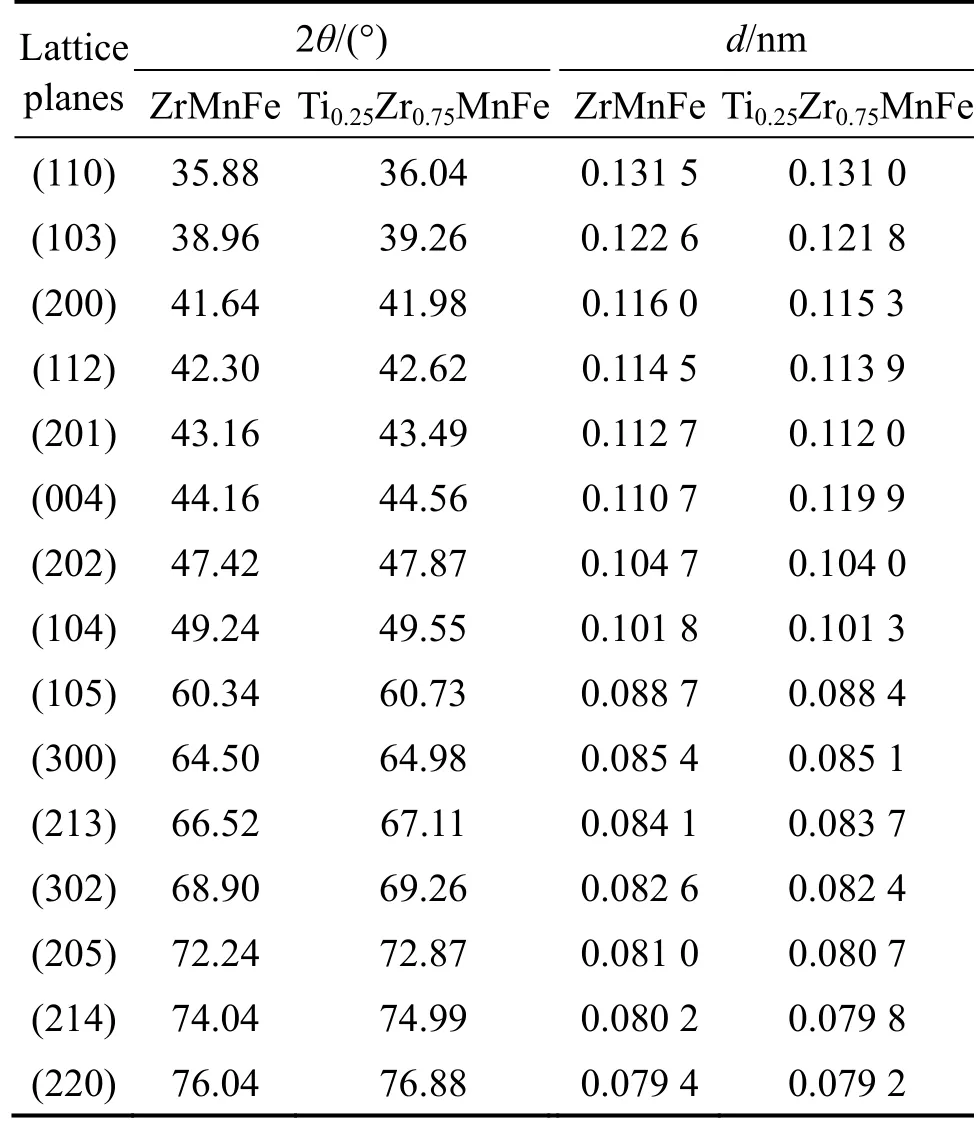
表1 TixZr1-xMnFe 合金各晶面的间距Table 1 Interplanar spacing of TixZr1-xMnFe alloys
图4 所示为ZrMnFe 合金过200 目筛后的SEM图及EDS 图谱。从图4(a)可知,粉末粒度基本在5~20 μm 之间,粒度较小较均匀。因为粒度较小的粉末表面积更大,吸附能力更强,因此ZrMnFe 合金有较强的CO2吸附力(低浓度中可以达到99.9%)。用 EDS 分析ZrMnFe 合金粉末的元素构成,结果表明合金表面Zr、Mn、Fe 元素分布很均匀。

图4 ZrMnFe 合金粉末的扫描电子显微镜照片(a)~(b)及能谱分析(c)~(d)Fig.4 SEM images (a)-(b) and EDS analyses (c)-(d) of ZrMnFe alloy powder
图5 所示为ZrMnFe 合金在660~700 ℃下的CO2吸附动力学曲线及吸附反应前后的样品实物图。从图5(a)发现,CO2的吸附量随时间延长而逐渐增加。起始8 h 内吸附速度较快,随后逐渐减慢直到吸附饱和,饱和吸附所需的时间约为164~168 h。

图5 ZrMnFe 合金在 660~700 ℃的CO2 吸附动力学曲线(a)以及反应前后实物图对比(b)Fig.5 CO2 adsorption kinetics curves (a) of ZrMnFe alloy at 660-700 ℃and comparison before and after reaction (b)
通过对比图5(a)不同温度下的吸附动力学曲线,结合表2 可知,随温度升高,ZrMnFe 合金对CO2的最终吸附量和起始8 h 内的吸附速率先升高后降低。680 ℃时,CO2的最终吸附量达到最高,为3.869 mmol/g,此时前8 h 有最大吸附速率0.301 mmol/(g·h),这表明ZrMnFe 合金对CO2的最佳吸附温度为680 ℃。从图5(b)看出,吸附前的样品为银色略带金属光泽,CO2饱和吸附后则变为乌黑。同时对比吸附前后样品的质量,结果如表2 所列,样品的质量增加率也呈先升高后降低的规律,680 ℃时达到最高值37.9%,这也证明680 ℃时吸附效果最好。
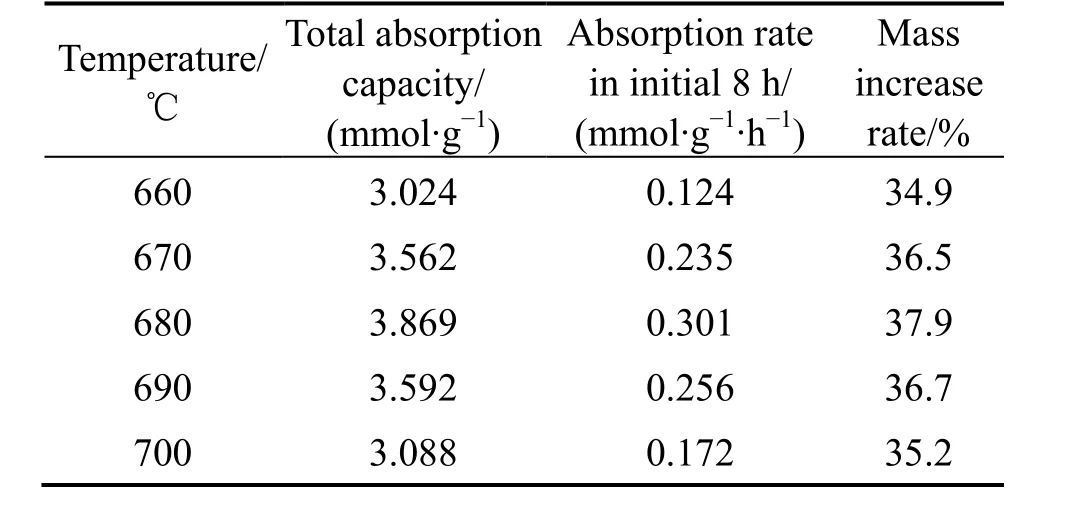
表2 660~700 ℃时ZrMnFe 合金CO2吸附量的相关数值Table 2 CO2 adsorption capacity, adsorption rate,and mass increase rate of ZrMnFe between 660-700 ℃
为了研究Ti 掺杂对ZrMnFe 基合金CO2吸附性能的影响,在最佳吸附温度680 ℃对TixZr1-xMnFe进行对比测试,结果如图6 所示。可以发现,Ti 掺杂后CO2吸附速率明显降低,但最终的吸附量提高。表3 所列为TixZr1-xMnFe 合金在 680 ℃的CO2吸附量、 吸附速率及样品的质量增加率。Ti0.25Zr0.75MnFe 的CO2最终吸附量为4.612 mmol/g,比ZrMnFe 提升了19.2%,样品的质量增加率为44.9%,比ZrMnFe 增加了18.5%。Ti 掺杂后反应速率明显降低,前8 h 的起始反应速率从0.301mmol/(g∙h)降低到0.119 mmol/(g∙h)。
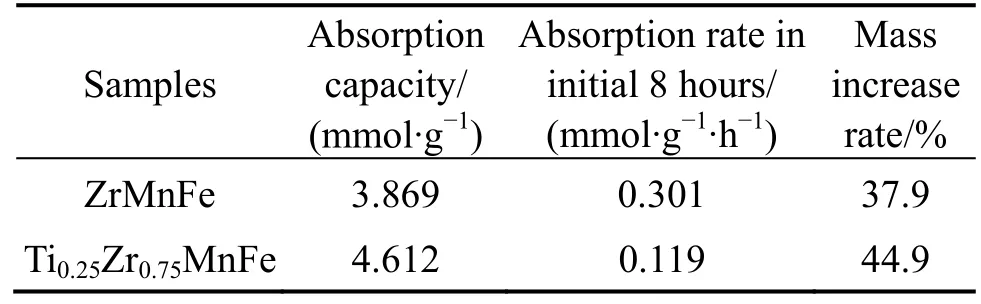
表3 680 ℃时TixZr1-xMnFe 合金CO2 吸附量的相关数值Table 3 CO2 adsorption capacity, absorption rate, and mass increase rate of TixZr1-xMnFe at 680 ℃
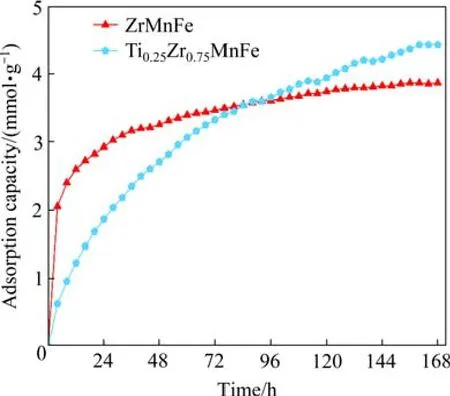
图6 TixZr1-xMnFe 合金在 680 ℃的CO2吸附动力学曲线Fig.6 CO2 adsorption kinetics curves of TixZr1-xMnFe alloys at 680 ℃
2.2 CO2 吸附后材料的结构
图7 和图8 所示分别为ZrMnFe 合金吸附CO2前后的XRD 图谱和SEM 图与EDS 能谱图。在ZrMnFe 合金对CO2气体的吸附过程中,合金与CO2发生反应,反应式为:Zr+CO2=ZrO2+C,从图7看出,经过168 h 的长时间吸附后,合金中原有的C14 Laves 相消失,取而代之的是新相ZrO2和C,这表明ZrMnFe 合金已完全与CO2气体发生反应。从图8 看出,合金表面存在O 和C 元素,表明ZrMnFe 合金成功吸附了CO2。

图7 CO2 吸附前后ZrMnFe 合金的XRD 图谱Fig.7 XRD patterns of ZrMnFe alloy before and after CO2 adsorption

图8 ZrMnFe 合金粉末吸附CO2 后的SEM 和EDS 图Fig.8 SEM images (a)-(b) and EDS spectra (c)-(d) of ZrMnFe alloy powder after CO2 adsorption
2.3 ZrMnFe 合金吸附CO2 的反应机理
实验所用CO2气体的成分如表4 所列(根据工业液体CO2的技术要求表可得)。ZrMnFe 合金吸附CO2发生反应生成ZrO2和C,通过检测反应容器内气体成分变化,可分析反应过程的机理。使用气体收集袋收集反应结束后的尾气和反应前的原始气体,通过气相色谱质谱联用仪检测分析气体成分[31]。

表4 实验所用CO2 气体成分Table 4 CO2 gas composition used in the experiment(mass fraction, %)
气相色谱质谱联用仪中的色谱仪具有分离色谱柱的能力,把物质按保留时间长短进行分离,结合质谱仪测定物质的质量与电荷的比值(m/Q),然后与标样保留时间进行对比,确定物质性质。图9 所示为初始气体和ZrMnFe 合金在680 ℃反应尾气的气相色谱分析,采集时间为0.2~3.7 min。可以发现初始气体有3 个色谱特征峰,反应尾气有2 个特征峰。表5 所列为5 个色谱特征峰对应的质谱处理分析,图10 所示为初始气体和ZrMnFe 合金在680 ℃反应尾气的特征峰相似物检索图。结合图10中各个特征峰相似物检索可以发现,初始气体中的特征峰1 与标准四羟基镍(nickel tetracarbonyl)的相似度指数(selectivity index, SI)为91%,推测该特征峰源于杂质,特征峰2 与标准CO2的SI 为91%,特征峰3 与标准水蒸气(Vapor)的SI 为98%,这符合表4 所列的CO2气体标准成分。而尾气中的特征峰4 与标准四羟基镍的SI 为93%,特征峰5 与标准CO2的SI 为90%,该特征峰的出现是由于当吸附达到饱和时,CO2气体不再被合金吸收,因此尾气也收集到了反应所用的CO2气体,这表明水蒸气被ZrMnFe 合金吸收,而四羟基镍没被吸收。尾气中并未检测出CO 或O2,说明CO2裂解反应发生后产生的氧原子和ZrMnFe 基体反应生成ZrO2等新相,且无中间产物CO 生成。

表5 色谱特征峰对应的质谱处理分析Table 5 Corresponding mass spectrometry analysis of chromatographic characteristic peak

图9 初始气体和ZrMnFe 合金在680 ℃反应尾气的气相色谱质谱分析图Fig.9 Gas chromatography-mass spectrometry of initial gas and reaction tail gas of ZrMnFe alloy at 680 ℃

图10 初始气体和ZrMnFe 合金在680 ℃反应尾气的特征峰与相似物的标准峰对比图Fig.10 Comparison between characteristic peak of initial and reaction tail gas of ZrMnFe alloy at 680 ℃ and standard peak of similar content
3 计算结果分析
3.1 CO2 分子在ZrMnFe (110)表面的吸附
图11 所示为ZrMnFe 的(110)表面与吸附位点。选取ZrMnFe (110)面5 个可能的高对称性吸附位点:表层Zr 原子的顶位(T1)、表层Fe 原子的顶位(T2)和桥位(B)、由4Zr1Fe 组成的空位(H1)和由1Zr2Fe组成的空位(H2)。考虑CO2气体分子的两种不同取向,如图12 所示,C—O—C 原子呈水平方向分布时,记为“Hor”; C—O—C 原子呈垂直方向分布时,记为“Ver”。将不同取向的CO2分子放置在ZrMnFe (110)表面的5 个吸附位点上方,构建模型如图13 所示,通过Materials-Studio 软件中的Castep 模块对初始模型进行优化。

图11 ZrMnFe 的(110) 清洁表面及各个吸附位点Fig.11 Top view of (110) clean surface and adsorption sites of ZrMnFe models

图12 不同CO2 结构Fig.12 Different orientations of CO2

图13 CO2 分子在ZrMnFe (110)表面的吸附位点Fig.13 Adsorption sites of CO2 molecule on the surface of ZrMnFe (110)
为了定量描述ZrMnFe (110)表面对CO2分子的吸附强度,计算吸附前后体系总能量的差值,即吸附能Ea,计算公式如下:
式中:Ea、EZrMnFe、ECO2和EZrMnFe/CO2分别表示吸附能、清洁的ZrMnFe (110)表面的能量、CO2分子的能量和ZrMnFe (110)表面吸附CO2分子体系的总能量。ZrMnFe (110)表面不同吸附位点对不同取向CO2气体的吸附能和吸附参数,如表6 所列。吸附能越大,表示样品吸附CO2后体系的能量越低,材料体系越稳定,因此越容易吸附。结果表明,最佳吸附位点为水平吸附的H1位点,吸附能为5.531 eV。此外水平吸附的所有吸附位点的吸附能都大于2.6 eV,而垂直吸附的所有吸附位点的吸附能均小于0.3 eV。

表6 CO2 分子吸附在ZrMnFe (110)表面的吸附能和O—C、C—O 键长Table 6 Adsorption energy and bond length of CO2 adsorbed on ZrMnFe (110) surface
表6 中列出了不同位点吸附的CO2分子的O—C 和C—O 键长。由于碳氧双键不易解离,可以发现垂直吸附的CO2分子的O—C 键与C—O 键键长变化不大,水平吸附的有一定的放松,其中Hor-H1位点吸附的CO2分子的O—C 和C—O 两个键分别从0.117 7 nm 放松到0.137 1 和0.310 9 nm,表明CO2分子在该位点易解离,吸附会变得更加容易,因此Hor-H1位点为CO2分子在ZrMnFe (110)表面的最佳吸附位点。
3.2 态密度分析
为了进一步分析CO2分子吸附后与ZrMnFe(110)表面原子之间的相互作用,分别计算CO2分子吸附在ZrMnFe (110)表面Hor-H1位点前后的态密度(density of states, DOS),结果如图14 所示。清洁表面和吸附后表面的总态密度(total density of states,TDOS)形状基本一致,都表现为金属性,但吸附CO2后在-21.68 eV 和-17.72 eV 处出现新的态密度峰,结合O 原子的偏态密度图(partial density of states,PDOS),可以推断这两个峰是由O 的2s 轨道提供。此外吸附CO2后Zr 原子的4p、4d 轨道在-21.68 eV和-17.72 eV 处也出现新的态密度峰,说明O 原子和Zr 原子发生轨道杂化,生成了O—Zr 共价键,此时Mn 和Fe 原子与O 原子之间并未发生轨道杂化。

图14 ZrMnFe (110)表面吸附CO2 分子前后的总态密度和分波态密度图Fig.14 Total and sub-wave density of states before (a) and after (b)-(c) CO2 adsorbed onto ZrMnFe (110) surface
3.3 CO2 分子在Ti0.25Zr0.75MnFe (110)表面的吸附
图15 所示为Ti0.25Zr0.75MnFe (110)表面7 个可能的高对称性吸附位点:表层Zr 原子的顶位(T1)、表层Fe 原子的顶位(T2)、表层Ti 原子的顶位(T3)、两个相邻Zr 原子中间的桥位(B1)、Zr 原子和的Ti原子中间的桥位(B2)、3Zr1Fe1Ti 组成的空位(H1)及1Zr2Fe 组成的空位(H2)。
CO2气体分子有两种不同取向,则共有14 种初始吸附位点,吸附位点的吸附能Eb计算公式如下:
式中:Eb、ETi0.25Zr0.75MnFe、ECO2和ETi0.25Zr0.75MnFe/CO2分别为吸附能、清洁的Ti0.25Zr0.75MnFe (110)表面能量、CO2分子的能量和Ti0.25Zr0.75MnFe (110)表面吸附CO2分子体系的总能量,计算结果列于表7。通过分析Ti0.25Zr0.75MnFe (110)表面不同吸附位点对不同取向CO2气体的吸附能可知,最佳吸附位点为Hor-T2,吸附能为4.945 eV,同时CO2分子的两个碳氧键分别从0.117 7 nm 放松到0.135 3 和0.308 1 nm,达到完全解离的状态;此外,Hor-H1、Hor-B1和 Hor-B2位点的吸附能分别为3.390、2.469 和3.734 eV,CO2分子的碳氧键也有一定的放松,达到部分解离;其余位点的吸附能均小于0.4 eV,CO2分子的碳氧键和自由CO2分子差距不大,处于未吸附的状态。
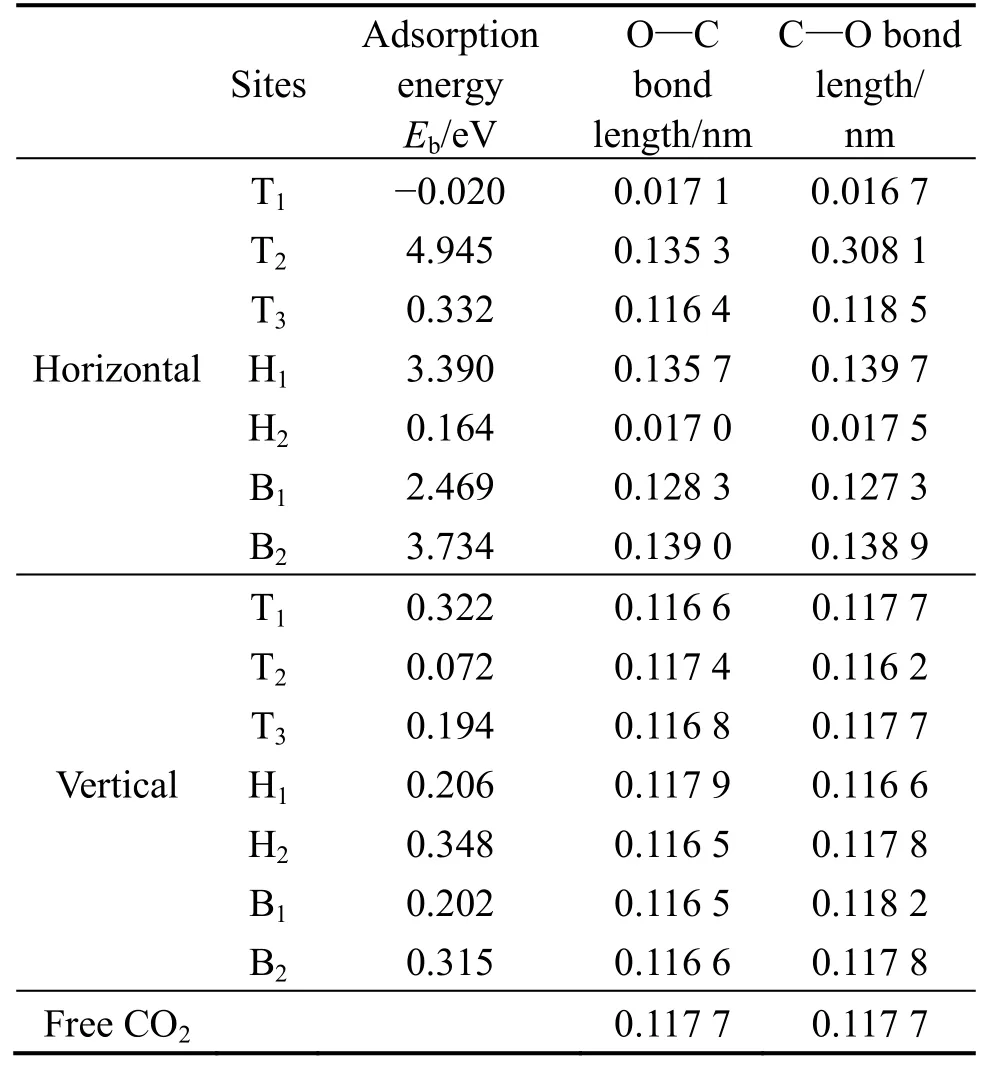
表7 CO2 分子吸附在Ti0.25Zr0.75MnFe (110)表面的吸附能及键长Table 7 Adsorption energy and bond length of CO2 adsorbed on Ti0.25Zr0.75MnFe (110) surface
对比表6 和表7 可知,CO2分子在ZrMnFe (110)表面的吸附能比在Ti0.25Zr0.75MnFe (110)表面的更大,CO2分子的解离程度也更大,这表明CO2分子在ZrMnFe (110)表面吸附性能更好。结合图6 中ZrMnFe 和Ti0.25Zr0.75MnFe 的CO2吸附实验结果可知,虽然经过 168 h 的长时间吸气测试后,Ti0.25Zr0.75MnFe 的最终吸附量优于ZrMnFe,但其前96 h 的吸附量比ZrMnFe 的低,且前8 h 的吸附速率更低,这表明前期CO2分子解离生成碳原子和氧原子的过程中,ZrMnFe 的能力优于Ti0.25Zr0.75-MnFe,和计算结果一致。
4 结论
1) 采用电弧熔炼-机械破碎-压制工艺制备的TixZr1-xMnFe(x=0、0.25) 吸气合金具有与ZrMn2相一致的C14 Laves 相,Ti 掺杂会导致晶面间距缩短。
2) 最终吸附CO2量随着温度升高先增大后减小,680 ℃时效果最好,此时,最终的CO2吸附量为3.869 mmol/g,前8 h 有最高吸附速率,为0.301 mmol/(g·h);Ti 掺杂后CO2的最终吸附量有所提升,为4.612 mmol/g,但前8 h 的吸附速率明显降低,为0.119 mmol/(g·h)。
3) ZrMnFe 合金粉末吸附CO2后,O 和C 元素多出现在粗糙表面的Zr 元素区域,ZrMnFe 合金和CO2反应生成新相ZrO2,CO2反应很彻底,且没有中间产物产生,也没有ZrMnFe 残留。
4) 第一性原理计算结果表明,CO2分子取向为水平时,ZrMnFe (110)表面对CO2分子的吸附能力较强,吸附能均在2.6 eV 以上,其中H1位点为最佳吸附位点,吸附能为5.531 eV。在CO2分子的吸附过程中,O 的2s 轨道和Zr 原子的4p、4d 轨道出现杂化,导致O 原子和Zr 原子的共价结合。CO2分子在Ti0.25Zr0.75MnFe (110)表面的最佳吸附位点为Hor-T2,吸附能为4.945 eV,略低于在ZrMnFe(110)表面的吸附能,CO2分子的碳氧键解离也弱于ZrMnFe (110)表面,说明CO2分子在Ti0.25Zr0.75MnFe(110)表面的解离性能较差,与实验中Ti0.25Zr0.75-MnFe 前96 h 的吸附性能差相对应。
猜你喜欢
山东陶瓷(2021年5期)2022-01-17
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
粉末冶金技术(2021年3期)2021-07-28
陶瓷学报(2021年1期)2021-04-13
中国有色金属学报(2018年2期)2018-03-26
中成药(2018年1期)2018-02-02
焊接(2016年8期)2016-02-27
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
