
2-(2-羟基苯亚甲基胺)-4,6-二羟基-嘧啶的质子转移异构化反应
2011-12-11 09:08周子彦苏忠民谢玉忠丁慎德王华静
物理化学学报 2011年9期
周子彦 刘 敏 苏忠民 谢玉忠 丁慎德 王华静
(1山东理工大学化学工程学院,山东淄博255049;2东北师范大学化学学院功能材料化学研究所,长春130024; 3延边大学理学院化学系,吉林延吉133002)
2-(2-羟基苯亚甲基胺)-4,6-二羟基-嘧啶的质子转移异构化反应
周子彦1,*刘 敏3苏忠民2谢玉忠3丁慎德1王华静1
(1山东理工大学化学工程学院,山东淄博255049;2东北师范大学化学学院功能材料化学研究所,长春130024;3延边大学理学院化学系,吉林延吉133002)
为了探索2-(2-羟基苯亚甲基胺)-4,6-二羟基-嘧啶(M1)分子醇式和酮式结构互变异构化的反应机理,利用密度泛函理论(DFT)方法,在B3LYP/6-311+G(d,p)基组水平上,对M1化合物异构化反应的势能面进行了研究,在探讨各种可能的反应途径中,发现单体至少有8种异构体和10种过渡态.结果表明:2-(2-羟基苯亚甲基胺)-6-羟基-4(3H)嘧啶酮(M6)不论是单体、与水形成的配合物,还是二聚体,比其相对应的异构体能量低,表明在通常情况下是以M6形式稳定存在的;在考察的可能反应途径中,直接进行的分子内质子转移过程需要的活化自由能为143.8 kJ·mol-1,水助催化时,反应的活化自由能为38.9 kJ·mol-1,二聚体双质子转移的活化自由能为0.6 kJ·mol-1,二聚体双质子转移所需活化自由能最低,在室温下就可以进行,由此可见氢键在降低反应活化能方面起着重要的作用.
2-(2-羟基苯亚甲基胺)-4,6-二羟基-嘧啶;密度泛函理论;互变异构;质子转移
1 引言
水杨醛缩芳胺类席夫碱由于具有化学性质稳定、结构特别易变的特殊性,对光、热、电、压等外界环境特别敏感,具有光致变色、非线性光学、光致发光等多种性质,此类化合物与众不同的分子内超快质子转移是实现电荷转移、传递的重要基础,因而引起了人们极大的研究兴趣.1,2质子转移而产生的互变异构现象,不但在一般化学研究中会发生,而且在分子生物学上也同样具有非常重要的意义.嘧啶碱基广泛地存在于生物体系中,质子在嘧啶碱基中杂原子间的迁移,主要是借助分子内氢键或者是溶剂中的氢键桥而发生的异构化反应,此过程导致嘧啶碱基有很多异构体,这些异构体的相对稳定性和异构体之间的相互转变对生物分子的活性具有重要影响.3,4随着量子化学和计算机技术的发展,质子转移和旋转异构反应都能被精确地计算.5-21我们曾经报道了8-羟基喹啉,22,236-甲基-4-羟基嘧啶24,25和水杨酰苯胺26质子转移的理论研究.但对水杨醛缩嘧啶胺席夫碱的研究报道却较少.27,282-(2-羟基苯亚甲基)胺基-4,6-二羟基-嘧啶分子内有一个O-H…N分子内氢键,存在着质子转移的通道.为了探讨其异构化反应的机理,我们利用量子化学的密度泛函理论对目标化合物的单体、水助催化和同种二聚体质子转移的异构化过程进行了研究,所研究的分子结构如图1所示,并标出了原子的序号.

图1 2-(2-羟基苯亚甲基)胺基-4,6-二羟基-嘧啶结构Fig.1 Structure of 2-(2-hydroxybenzylidenamino) pyrimidine)-4,6-diol
2 计算方法
利用DFT方法,在B3LYP/6-311+G(d,p)水平上,对标题化合物的单体质子转移、水助催化质子转移和同种二聚体质子转移异构化过程进行了研究;在相同水平下进行了振动分析,确认了异构体的稳定态和一阶鞍点过渡态;并通过内禀反应坐标分析(IRC)对异构体之间对应的过渡态进行了确认;全部计算用Gaussian 03量子化学程序包29完成.
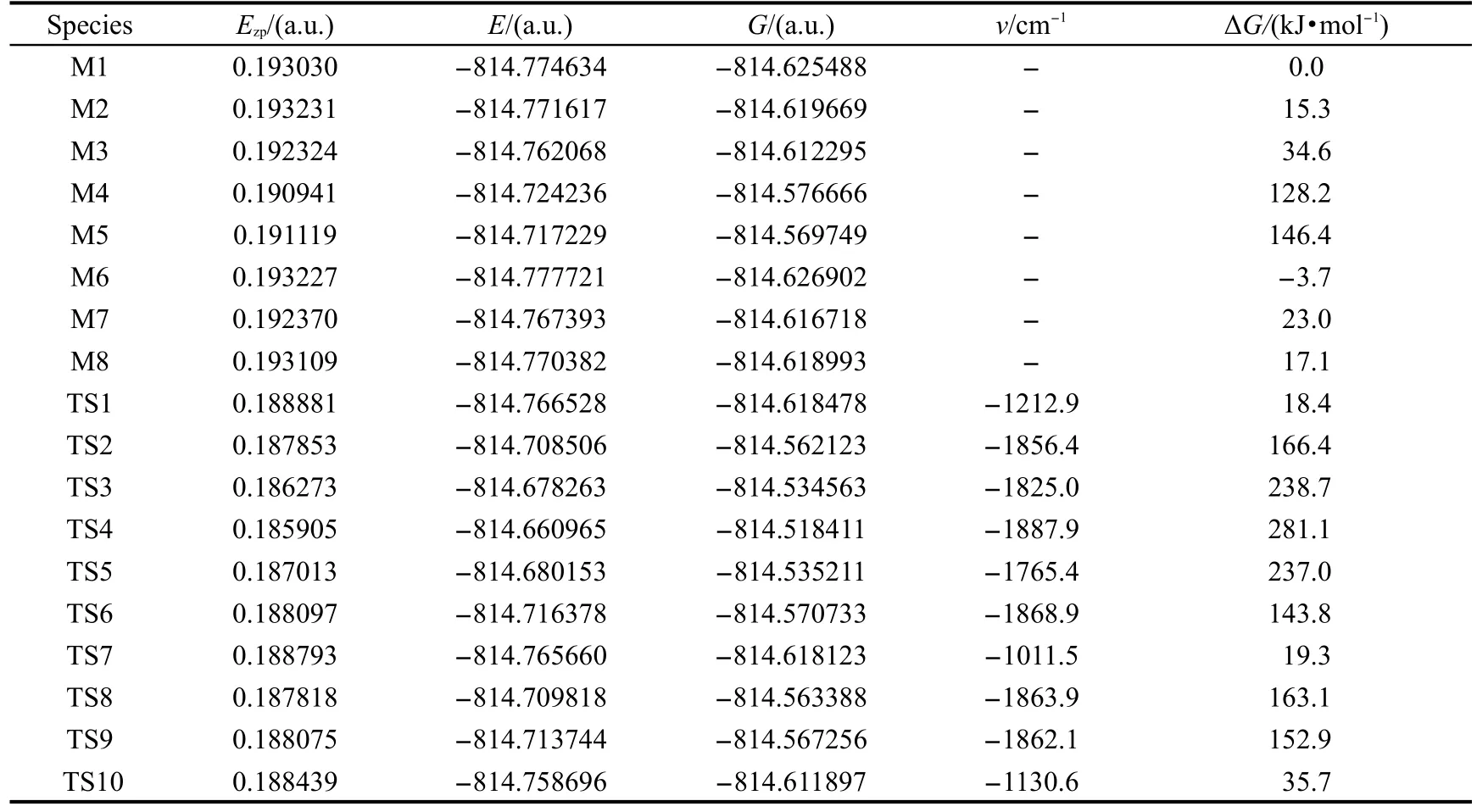
表1 DFT-B3LYP/6-311+G(d,p)方法计算得到的目标分子单体各异构体和过渡态的零点能(Ezp)、总能量(E)、吉布斯自由能(G)及谐振频率vTable 1 Zero-point energy(Ezp),total energy(E),Gibbs free energy(G)and imaginary frequency(ν)of various species isomers and transition states in reaction pathways by intra molecular prototropy obtained with the DFT-B3LYP/6-311+G(d,p)calculations
3 结果与讨论
3.1 2-(2-羟基苯亚甲基)胺基-4,6-二羟基-嘧啶的分子内质子转移过程
图2给出了标题化合物单体所有反应途径的各种异构体和过渡态.在DFT-B3LYP/6-311+G(d,p)水平下计算得到的零点能、总能量、吉布斯自由能和振动频率列在表1中.反应势能面及能垒见图3.图4给出了M1→M2,M1→M6沿反应路径异构化过程的几何构型部分参数.
计算结果表明,目标分子基态时的所有异构体的能量次序为M5>M4>M3>M7>M8>M2>M1>M6. M6是最稳定的构型,其次是醇式构型M1.除M4和M8外,所有构型几乎都是呈平面型的.在M4中嘧啶环与苯环之间的二面角为151.2°,H(20)与嘧啶环的二面角为20.5°,而M8中嘧啶环与苯环之间的二面角为142.4°.除了M4和M5外,在其他构型中分别存在有不同的O(15)-H(21)…N(7)和O(15)…H(21)-N(7)分子内氢键作用,例如:M1和M3中的O(15)-H(21)…N(7)氢键的键长分别为0.1722和0.1740 nm,M2和M6中的O(15)…H(21)-N(7)氢键键长分别为0.1780和0.1769 nm,而M4和M5由于缺少分子内氢键的作用,所以两者的能量均比其他异构体高.

图2 目标分子基态分子内质子转移反应途径中的异构体和过渡态Fig.2 Geometric structures of isomers and transition states of 2-(2-hydroxybenzylidenamino)pyrimidine)-4,6-diol in reaction pathways via intramolecular prototropy

图3 DFT-B3LYP/6-311+G(d,p)方法计算得到的标题分子基态的质子转移异构化反应势能面Fig.3 Potential energy surface obtained with the DFT-B3LYP/6-311+G(d,p)calculations for title moleculeM1 is taken as zero point.
如图3所示,我们找到了4条从醇式构型M1异构化为酮式构型M6的反应路径.它们分别是:
(1)M1→TS6→M6;
(2)M1→TS1→M2→TS8→M7→TS7→M6;
(3)M1→TS9→M8→TS10→M3→TS2→M2→ TS8→M7→TS7→M6;
(4)M1→TS1→M2→TS2→M3→TS3→M4→TS4→M5→TS5→M2→TS8→M7→TS7→M6.
在四个反应路径中最高的反应活化自由能分别为143.8、163.1、166.4和281.1 kJ·mol-1.路径(1)中从M1→M6是放热反应,放出热量约为3.7 kJ· mol-1,但从M1→M6的活化自由能垒较高,达到了143.8 kJ·mol-1,在常温下很难进行.路径(2),(3)和(4)中,同样由于较高的活化自由能也很难直接进行分子内质子转移.
图4表示的是M1→TS1→M2(A)和M1→TS6→M6(B)异构化过程中经IRC分析的分子关键部位的几何参数变化.对于图4A,随着反应开始,O(15)-H(21)键逐渐伸长,N(7)-H(21)键逐渐缩短,∠O(15)-H(21)-N(7)逐渐增大形成了稳定的六元环过渡态,随着反应的进行,H(21)转移至N(7)上, O(15)-H(21)键长进一步伸长,N(7)-H(21)键长进一步缩短形成酮式构型M2,∠O(15)-H(21)-N(7)也逐渐的变小直至稳定.对于B图,随着反应从醇式构型M1开始,O(17)-H(20)键逐渐伸长,N(1)-H(20)键逐渐缩短,∠O(17)-H(20)-N(1)逐渐增大形成了四元环过渡态,随着反应的进行,H(20)转移至N1上,O(17)-H(20)键长进一步伸长,N(1)-H(20)键长进一步缩短,∠O(17)-H(20)-N(1)也逐渐的变小直至形成稳定的M6.因为M1→TS6→M6形成的过渡态是四元环,而M1→TS1→M2形成的过渡态是六元环,四元环的张力较六元环大,所以这也就可以得出M1与M6互变异构反应的活化自由能比M1与M2互变异构反应的活化自由能高的结果.

图4 DFT-B3LYP/6-311+G(d,p)方法计算得到的M1→TS1→M2和M1→TS6→M6基态异构化过程中经IRC分析的分子关键部位的几何参数变化Fig.4 Variation of geometric configuration parameters of M1→TS1→M2 and M1→TS6→M6 in the isomerization reaction pathway obtained by intrinsic reaction coordination analysis with the DFT-B3LYP/6-311+G(d,p)calculations
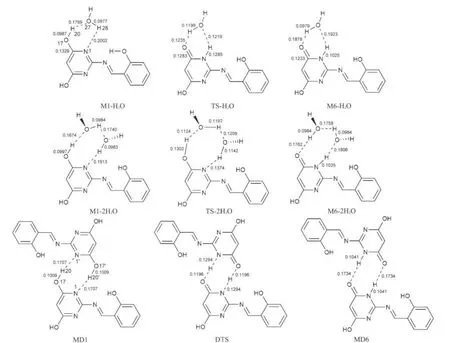
图5 DFT-B3LYP/6-311+G(d,p)方法计算得到的水助催化质子转移和二聚体双质子转移过程的异构体和过渡态构型Fig.5 Geometric structures of isomers and transition states of object compound with water and its dimmers in reaction pathways via intra molecular prototropy obtained with the DFT-B3LYP/6-311+G(d,p)calculations bond length in nm
3.2 水助催化质子转移过程(M-H2O,M-2H2O)和二聚体(MD)双质子转移过程
从表1中发现M6构型在所有的异构体中是最稳定的,但是直接与M1进行异构化反应所需的活化自由能较高,常温下直接进行分子内的质子转移比较困难.所以我们另外设计了水助催化质子转移和二聚体双质子转移过程,并在DFT/B3LYP/6-311+ G(d,p)水平上对其进行了研究.
图5给出了水助催化质子转移过程和二聚体双质子转移过程的异构体和过渡态构型,主要键长参数均标注于图中.水合物、二聚体的异构体和过渡态的零点能、总能量、吉布斯自由能和谐振频率见表2.
从图5中可看出,这两种途径的质子转移过程均发生在平面内.在这两种反应途径中出现了3种类型的氢键分别是O-H…O,O-H…N和N-H…O.水助催化质子转移过程中,一水分子参与催化时,水中的H和O原子参与形成了一个六元环,而两水分子参与催化时,水中的H和O原子参与形成的是一个八元环,在二聚体双质子转移过程中,形成的也是一个八元环.图5列出了这三种氢键的键长计算值,氢键的键长越短,键角越接近180°,氢键的强度也就越大.30对于二聚体双质子转移过程中的O-H…N和O…H-N氢键键长均短于水助催化质子转移过程的相应值,而前者的氢键键角几乎接近直线,所以二聚体双质子转移过程中的氢键强度大于水助催化质子转移过程的氢键强度.在水助催化质子转移过程中,二水配合物中的O-H…N和O…H-N氢键键长比一水配合物中的短,氢键的强度强.在单体M1中,O-H键长为0.0968 nm,振动波数为3766.0 cm-1,形成复合物M1-H2O、M1-2H2O和二聚体DM1后,O-H键长分别增长为0.0989、0.0997和0.1009 nm,振动波数分别降至3374.7、3181.1和2982.7 cm-1,O-H键的伸缩振动表现为红移情况,所以O-H…N为红移型氢键.31单体M6中N-H键长为0.1013 nm,振动波数为3592.4 cm-1.形成复合物M6-H2O,M6-2H2O和二聚体DM6之后, N-H键长分别也增长为0.1025、0.1035和0.1041 nm,振动频率分别也降低至3373.5、3195.3和3103.0 cm-1,N-H键的伸缩振动也表现为红移情况,NH…O为红移型氢键.同样,O-H…O氢键也为红移型氢键.
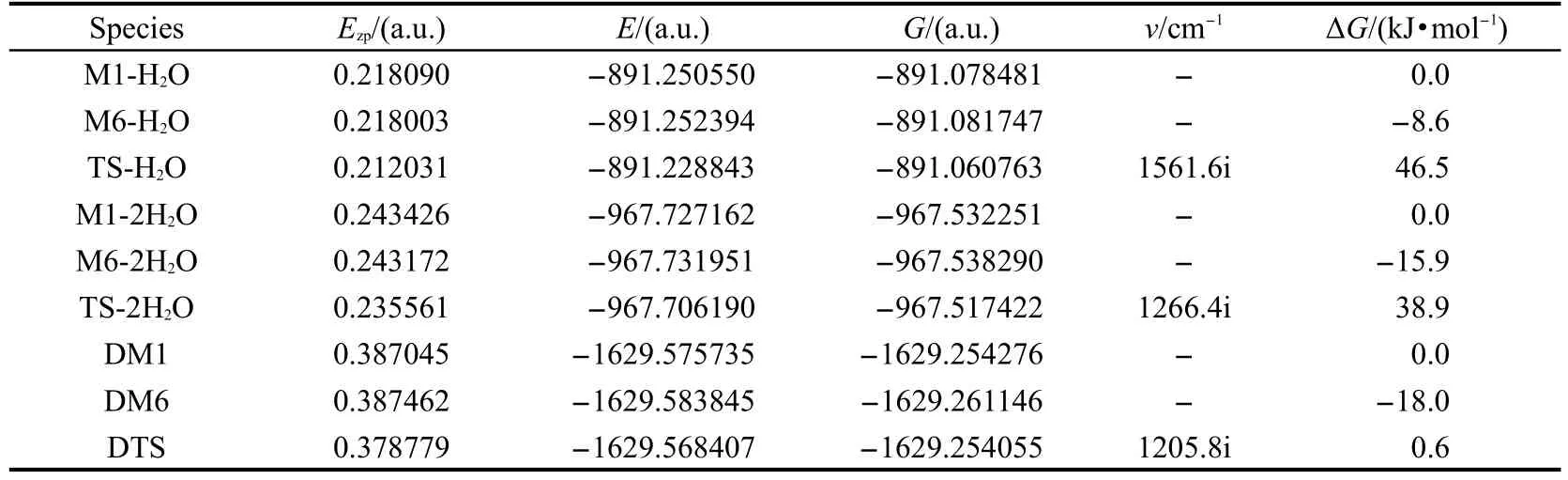
表2 DFT-B3LYP/6-311+G(d,p)方法计算得到的水配合物和二聚体各异构体和过渡态的零点能(Ezp)、总能量(E)、吉布斯自由能(G)及谐振频率(v)Table 2 Zero-point energy(Ezp),total energy(E),Gibbs free energy(G)and imaginary frequency(ν)of various species isomers and transition states in reaction pathways of water complex and dimer obtained with the DFT-B3LYP/6-311+G(d,p)calculations
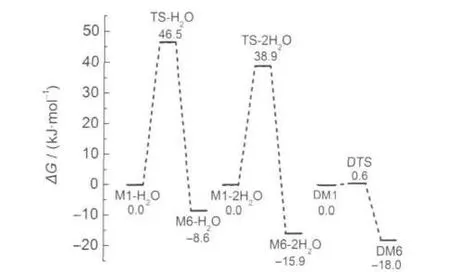
图6 DFT-B3LYP/6-311+G(d,p)方法计算得到的比较2种反应途径能量(kJ·mol-1)变化的示意图Fig.6 Schematic diagram of potential energy(kJ·mol-1) surface of comparing with two reaction pathways obtained with the DFT-B3LYP/6-311+G(d,p)calculationsReaction matters energy is taken as zero point.
表2列出了各物种的零点能、总能量、吉布斯自由能及谐振频率的计算结果.从中发现酮式二聚体DM6的能量最低,与醇式异构体相比稳定.图6给出了2种反应途径的势能面曲线.从图2和图6可以看出,由醇式变为酮式结构的所有反应均是放热反应,单体M1→M6直接进行分子内的质子转移,翻越的活化自由能垒是143.8 kJ·mol-1,如此高的活化自由能在常温下是很难进行的,其主要原因是在质子转移过程中没有氢键的参与,并且在形成过渡态的过程中,形成了一个具有较大张力的四元环,以致这一过程的活化自由能较高.当水作为催化剂参与反应后,所需要的活化自由能就大幅降低,一水参与反应的活化自由能为46.5 kJ·mol-1,二水参与反应的活化自由能为38.9 kJ·mol-1,这是因为在此过程中,前者形成了一个六元环的过渡态,后者形成了一个八元环的过渡态,张力较四元环小,二水通过八元环参与质子传递比一水通过六元环参与质子传递更为有效,32-34同时在整个过程中氢键OH…O,O-H…N,O…H-N也参与了反应.与前两种反应途径相比,二聚体双质子转移过程的活化自由能更低,所需反应活化自由能仅为0.6 kJ·mol-1,逆反应活化自由能也只有18.6 kJ·mol-1,因此常温下二聚体的质子转移很容易进行.这是由于二聚体质子转移过程形成了一个八元环的过渡态,分子张力比四元环和六元环都小;同时又由于O-H…N和O…H-N氢键的作用,并且它的氢键强度35比水助催化过程的大,使得二聚体异构体更稳定,发生双分子间的质子转移活化自由能降低.根据单体和二聚体的总能量可求得O-H…N和N-H…O氢键的键能31分别为66.9和71.9 kJ·mol-1,说明二聚体的分子间氢键强度大,二聚体分子更加稳定.另外,由于N-H…O键能比O-H…N键能大,同时也就说明了酮式二聚体DM6比醇式的DM1更稳定.
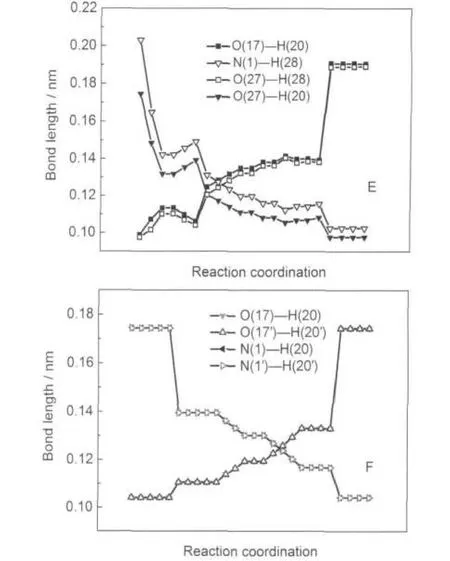
图7 DFT-B3LYP/6-311+G(d,p)方法计算得到的水助催化(E)和二聚体(F)反应路径的分子关键部位的几何构型参数变化的内禀反应坐标(IRC)分析Fig.7 Variation of geometric configuration parameters of catalysis water(E)and dimers(F)in the isomerization reaction pathway obtained by intrinsic reaction coordination analysis with the DFT-B3LYP/6-311+ G(d,p)calculations
图7中的E是水助催化质子转移异构化反应过程中经IRC分析的分子关键部位的几何参数变化,随着反应进行,M1-H2O中的O(27)-H(20)和N(1)-H(28)键逐渐减小,O(17)-H(20)和O(27)-H(28)键逐渐增长直至形成六元环过渡态,反应进一步进行,H(28)转移至N(1),直至形成M6-H2O.F为二聚体分子异构化过程中经IRC分析的分子的关键部位几何参数变化,随着反应的进行,O(17)-H(20)和O(17ʹ)-H(20ʹ)键逐渐增长,N(1)-H(20)和N(1ʹ)-H(20ʹ)键逐渐缩短,图中曲线O(17)-H(20)和O(17ʹ)-H(20ʹ),N(1)-H(20)和N(1ʹ)-H(20ʹ)重叠,说明二聚体质子转移过程是一个协同质子转移的过程.
4 结论
(1)计算结果表明,2-(2-羟基苯亚甲基)胺基-6-羟基-4(3H)嘧啶酮(M6)不论是单体,与水形成配合物,还是二聚体,与其相应的异构体比能量是低的,在通常情况下是以M6形式稳定存在的.
(2)通过对三种可能反应机理的研究表明,直接进行分子内质子转移过程需要最大的活化自由能143.8 kJ·mol-1,当水参与反应后,活化自由能降低到38.9 kJ·mol-1,二聚体分子间双质子转移的活化自由能进一步降低到0.6 kJ·mol-1,反应在室温下就可以快速进行.
(3)在反应过程中,氢键起着非常重要的作用,它的形成大幅度降低了反应的活化能.氢键的形成是水助催化质子转移和二聚体分子间双质子转移活化能降低的重要原因.
(4)在二聚体分子间异构化反应中,两个氢的迁移是同步进行的;而在水助催化间异构化反应中,两个氢的迁移不是同步进行的.
(1)Hadjoudis,E.;Dziembowska,T.;Rozwadowski,Z.J.Photoch. Photobio.A 1999,128,97.
(2) Kunkely,H.;Vogler,A.J.Photoch.Photobio.A 2001,138,51.
(3) Gazivoda,T.;Plevnik,M.;Plavec,J.;Kraljević,S.;Kralj,M.; Pavelić,K.;Balzarini,J.;De Clercq,E.;Mintas,M.;Raić-Malić,S.Bio.Med.Chem.2005,13(1),131.
(4)Huang,Y.Q.;Kenttämaa,H.J.Phys.Chem.A 2003,107(24), 4893.
(5)Peng,H.L.;Yu,X.Y.;Yi,P.G.;Wang,Z.X.;Li,X.F.;Wang, T.;Zhou,J.M.Acta Phys.-Chim.Sin.2010,26(1),141.[彭洪亮,于贤勇,易平贵,汪朝旭,李筱芳,王 涛,周继明.物理化学学报,2010,26(1),141.]
(6) Shi,J.Y.;Dong,L.H.;Liu,Y.J.Acta Phys.-Chim.Sin.2010,26 (12),3329.[史俊友,董丽花,刘永军.物理化学学报,2010, 26(12),3329.]
(7) Xu,X.F.;Gao,F.;Li,H.R.;Zhang,S.T.Acta Phys.-Chim.Sin. 2010,26(1),131.[徐晓芳,高 芳,李红茹,张胜涛.物理化学学报,2010,26(1),131.]
(8)Wang,L.X.;Yi,D.H.;Zou,H.T.;Xu,J.;Xu,W.L.Acta Phys.-Chim.Sin.2010,26(1),149.[王罗新,易第海,邹汉涛,许 杰,徐卫林.物理化学学报,2010,26(1),149.]
(9) Liu,Y.H.;Mehata,M.S.;Liu,J.Y.J.Phys.Chem.A 2011,115, 19.
(10)Zhao,X.;Chen,M.J.Phys.Chem.A 2010,114,7786.
(11) Moser,A.;Range,K.;York,D.M.J.Phys.Chem.B 2010,114, 13911.
(12) Manoj,K.N.J.Photoch.Photobio.A 2011,217,40.
(13) Li,A.;Yan,T.;Shen,P.J.Power Sources 2011,196,905.
(14) Xia,S.W.;Xu,X.;Sun,Y.L.;Fan,Y.H.;Bi,C.F.;Zhang,D. M.;Yang,L.R.Chin.J.Struct.Chem.2006,25,197.
(15)Abdel-Latif,S.A.;Hassib,H.B.;Issa,Y.M.Spectrochim Acta A 2007,67,950.
(16) Yu,W.;Yang,L.;Zhang,T.L.;Zhang,J.G.;Ren,F.J.;Liu,Y. H.;Wu,R.F.;Guo,J.Y.J.Mol.Struct.2006,794,255.
(17) Lang,H.Y.;Cai,J.;Shen,Y.H.;Jia,Y.Q.Acta Chimica Sinica 2005,63,814.[郎惠云,蔡 健,申烨华,贾璎琦.化学学报, 2005,63,814.]
(18) Koll,A.;Karpfen,A.;Wolschann,P.J.Mol.Struct.2006,790, 55.
(19)Xiao,F.R.;Chen,L.;Wang,J.D.;Wu,R.L.;Yue,F.;Li,J.Acta Chimica Sinica 2006,64,1517.[肖芙蓉,陈 鹭,王吉德,武荣兰,岳 凡,李 静.化学学报,2006,64,1517.]
(20) Makhmutova,R.I.;Vakulin,I.V.;Talipov,R.F.;Movsumzade, E.M.;Chuvashov,D.A.J.Mol.Struct.-Theochem 2007,819, 21.
(21)Kołodziej,B.;Grech,E.;Schilf,W.;Kamienski,B.;Makowski, M.;Rozwadowski,Z.;Dziembowska,T.J.Mol.Struct.2007, 32,844.
(22) Zhao,J.Y.;Zhou,Z.Y.;Su,Z.M.;Xie,Y.Z.;Sun,G.G.;Wu, X.Chin.J.Chem.2006,24(6),724.
(23) Zhou,Z.Y.;Shan,G.G.;Zhu,Y.L.;Yu,X.J.;Dong,Y.H.; Zhao,J.Y.J.Struct.Chem.2009,50(4),606.
(24)Zhou,Z.Y.;Zhao,J.Y.;Liu,M.;Su,Z.M.;Sun,G.Y.Chem.J. Chin.Univ.2007,28(12),2385. [周子彦,赵继阳,刘 敏,苏忠民,孙光延.高等学校化学学报,2007,28(12),2385.]
(25)Zhao,J.Y.;Zhou,Z.Y.;Su,Z.M.;Xie,Y.Z.;Wu,X.Chem.J. Chin.Univ.2006,27(3),494.[赵继阳,周子彦,苏忠民,谢玉忠,吴 学.高等学校化学学报,2006,27(3),494.]
(26)Zhou,Z.Y.;Shan,G.G.;Liao,Y.;Xing,W.;Yang,S.Y.;Su,Z. M.J.Mol.Struct.-Theochem 2010,945,110.
(27) Cherayath,S.;Alice J.;Chathakudam,P.P.Transition Metal Chem.1990,15,449.
(28)Francisco,H.U.;Nuria,A.I.C.;Miguel,N.M.C.;Jose,M.M. M.;Maria,J.R.E.J.Inorg.Biochem.2003,94,326.
(29) Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03, Revision B.03;Gaussian Inc:Pittsburgh,PA,2003.
(30) Chen,W.K.;Xu,J.;Zhang,Y.F.;Zhou,L.X.;Li,J.J.Acta Phys.-Chim.Sin.2002,18,802. [陈文凯,许 娇,章永凡,周立新,李俊篯.物理化学学报,2002,18,802.]
(31) Li,Q.;Huang,F.Q.Acta Phys.-Chim.Sin.2005,21,52. [李 权,黄方千.物理化学学报,2005,21,52.]
(32)Sun,X.M.;Wei,X.G,.;Wu,X.P.;Ren,Y.;Wong,N.B.;Li,W. K.J.Phys.Chem.A 2010,114(1),595.
(33)Sun,X.M.;Wei,X.G.;Wu,X.P.;Ren,Y.;Wong,N.B.;Li,W. K.Theor.Chem.Acc.2010,127,493.
(34)Deng,C.;Wu,X.P.;Sun,X.M.;Ren,Y.;Sheng,Y.H. J.Comput.Chem.2009,30,285.
(35) Jin,H.W.;Feng,J.K.;Ren,A.M.;Li,Z.R.;Wang,Z.Q.; Zhang,X.Acta Chimica Sinica 2000,58,194.[金宏威,封继康,任爱民,李志儒,王志强,张 希.化学学报,2000,58, 194.]
February 28,2011;Revised:May 26,2011;Published on Web:July 4,2011.
Proton-Transfer Isomerization Reactions of 2-(2-Hydroxybenzylidenamino)pyrimidine-4,6-diol
ZHOU Zi-Yan1,*LIU Min3SU Zhong-Min2XIE Yu-Zhong3DING Shen-De1WANG Hua-Jing1
(1College of Chemical Engineering,Shandong University of Technology,Zibo 255049,Shandong Province,P.R.China;2Institute of Functional Material Chemistry,Faculty of Chemistry,Northeast Normal University,Changchun 130024,P.R.China;3Department of Chemistry,College of Science,Yanbian University,Yanji 133002,Jilin Province,P.R.China)
To determine the tautomerism mechanism between the enol form and the keto form of 2-(2-hydroxybenzylidenamino)pyrimidine-4,6-diol(M1)the potential energy surface of the isomerization was studied using density functional theory(DFT)calculations at the B3LYP/6-311+G(d,p)level.We found that there were at least 8 isomers and 10 transition states in the possible reaction pathways.All the possible processes of the reaction were studied.The results showed that the energy of 6-hydroxy-2-(2-hydroxybenzylideneamino)pyrimidine-4(3H)-one(M6)was lower than those of the other isomers in the form of a monomer,a hydrate,and a dimer.Therefore,it was the most stable isomer.In these possible reaction pathways the activation free energy required for intramolecular prototropy was 143.8 kJ·mol-1and for the proton transfer process that was catalyzed by water was 38.9 kJ·mol-1.The activation free energy in the double-proton transfer of the dimer was 0.6 kJ·mol-1,which was the lowest value.The latter pathway was feasible at room temperature.This implies that hydrogen bonding plays an important role in depressing the activation energy of the reaction.
2-(2-Hydroxybenzylidenamino)pyrimidine)-4,6-diol;Density functional theory; Tautomerism;Proton transfer
∗Corresponding author.Email:zyzhou@sdut.edu.cn;Tel:+86-533-2767606.
The project was supported by the National Natural Science Foundation of China(20703008)and Natural Science Foundation of Shandong Province, China(ZR2009BL024).
国家自然科学基金(20703008)和山东省自然科学基金(ZR2009BL024)资助项目
O641
猜你喜欢
系统仿真技术(2022年4期)2023-01-17
北京航空航天大学学报(2022年5期)2022-06-06
浙江化工(2022年4期)2022-05-07
石油石化绿色低碳(2020年2期)2020-12-31
石油石化绿色低碳(2019年6期)2019-01-14
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
国外医药(抗生素分册)(2016年4期)2016-07-12
信息记录材料(2016年4期)2016-03-11
