
舌鳎亚科鱼类线粒体控制区快速进化及其重复序列延伸机制初步研究*
2012-01-08 08:17孔晓瑜江金霞苗宪广
中国海洋大学学报(自然科学版) 2012年1期
时 伟,孔晓瑜,江金霞,苗宪广
(1.中国海洋大学水产学院,山东青岛266003;2.中国科学院南海海洋研究所海洋生物资源可持续利用重点实验室热带海洋生物标本馆,广东广州510301)
舌鳎亚科鱼类线粒体控制区快速进化及其重复序列延伸机制初步研究*
时 伟1,2,孔晓瑜2**,江金霞2,苗宪广2
(1.中国海洋大学水产学院,山东青岛266003;2.中国科学院南海海洋研究所海洋生物资源可持续利用重点实验室热带海洋生物标本馆,广东广州510301)
本研究测定双线舌鳎Cynoglossus bilineatus(Lacepède,1802)、长钩须鳎Paraplagusia bilineata(Bloch,1787)和短钩须鳎Paraplagusia blochii(Bleeker,1851)3种舌鳎亚科鱼类的线粒体(mtDNA)控制区全序列,并与舌鳎亚科其它4种及鲽形目其他科的5种鱼类mtDNA控制区结构和序列进行了比较分析。结果显示,3种舌鳎鱼类的线粒体控制区全长从655~1 155bp,长度差异明显;舌鳎亚科鱼类控制区结构与其他鲽形目鱼类相似性很低,保守区系列中只能识别出CSB-A和TAS。碱基组成比较发现这3种舌鳎鱼类的AT含量比鲽形目其它鱼类要高。分析认为上述事件可能都是由该类群鱼类控制区的快速进化导致的。所有3种舌鳎的控制区在5’端都存在串联重复序列,每个重复单元中都存在类似于TAS的相关序列,并且重复序列可以形成非常稳定的二级结构。这2种特征为形成线粒体重复序列的非正常延伸机制(Illegitimate elongation model)提供了新的证据,同时,推测了这些舌鳎鱼类重复序列可能的延伸过程。
鲽形目;舌鳎亚科;控制区;重复序列;非正常延伸
舌鳎亚科(Cynoglossinae)隶属于鲽形目(Pleuronectiformes)、鳎超科(Soleoidei)、舌鳎科(Cynoglossidae)。舌鳎科鱼类大多为底层海水鱼类,根据Nelson[1]2006年统计认为世界范围内舌鳎科共有3属127种,其中舌鳎亚科大约有50种左右。李思忠等[2]记录了中国海域舌鳎科共3属约32种,其中舌鳎亚科有29种。舌鳎亚科主要分布于西太平洋及印度洋热带及温带泥沙底海区,在温带随着纬度升高种类减少;只少数能进入淡水,极少终生生活在淡水中。在我国,舌鳎亚科广泛分布于南海、东海和黄海水域。
脊椎动物线粒体基因组大小约为15~20kb的双链环状DNA分子,通常含有13种蛋白质编码基因、2种rRNA和22种tRNA,共计37个基因[3]。线粒体DNA(mt DNA)不同区域进化速率相差很大,一般来说,线粒体基因组中的控制区(CR)进化速率最快[4],由于其不编码蛋白质等原因,从而受到较小的选择压力,能迅速积累了较多的突变,如碱基替换、插入、缺失以及众多的串联重复序列等。尽管控制区进化速度快,但是其中一般都存在3个保守区域:终止相关序列区(Extended termination associated sequence domain,ETAS)、中央保守区(Central conserved sequence blocks domain,CSB A-F)和保守序列区(Conserved sequence blocks domain,CSB 1-3)[5-6]。控制区的两端经常会出现串联重复序列[7-8],关于重复序列产生机制存在多种假说,如分子内分子间重组(intra-or intermolecular recombination)[9-11]、滑链错配(slippedstrand mispairing)[12]、非正常延伸(illegitimate elongation model)[13]等。
在测定的半滑舌鳎(Cynoglossus semilaevis)的线粒体全序列中发现其线粒体基因组存在基因重排和易位现象[14],控制区也从正常的tRNAPro和tRNAPhe之间移位到ND1基因和tRNAGln之间,并且序列发生非常显著地变异,其序列和同1个亚目的鳎科鱼类塞内加尔鳎(Solea senegalensis)[15]控制区相似性极低,和其它硬骨鱼类则基本没有相似性。在亲缘关系十分相近的2个科内,存在如此巨大的差异是十分罕见的,在硬骨鱼类中还没有报道过。
为了探讨线粒体控制区的重排是否稳定存在于舌鳎亚科,本文测定了舌鳎亚科的双线舌鳎Cynoglossus bilineatus(Lacepède,1802)、长钩须鳎Paraplagusia bilineata(Bloch,1787)和短钩须鳎Paraplagusia blochii(Bleeker,1851)线粒体控制区全序列,以期找到这种快速变异的原因。同时研究了舌鳎亚科鱼类控制区的串联重复序列可能的产生及延伸过程,并且对重复序列折叠的二级结构功能进行了推测。
1 材料方法
1.1 材料
本实验用双线舌鳎、长钩须鳎取自湛江,短钩须鳎样品取自阳江,每种鱼取1个样品,取肌肉组织-20℃保存。采用OMEGA肌肉组织提取试剂盒提取的总DNA,通过1%的琼脂糖电泳对提取的效果进行检测。并从GenBank下载已有的鲽形目其它鱼类控制区全序列(见表1)。

表1 用于本研究的鲽形目鱼类线粒体控制区的相关信息Table 1 The information of mitochondrial control regions of flatfishes in this study
1.2 方法
用PCR扩增和直接测序法测定线粒体控制区全序列。根据NCBI核酸数据库上已有的舌鳎类线粒体控制区序列设计2对引物进行PCR扩增,Z-2733:ATCCAGGTCAGTTTCTATC、F-5196:CTAAATGGTTGGGGTATGG、Pleur-Z2625:GTTTACGACCTCGATGTTGGATCAGGACAT、Pleur-F6746:GCGGTGGATTGTAGACCCATARACAGAGGT,PCR反应总体积为25μL:2.0mmol/L MgCl2、0.2mmol/L dNTP、0.5μmol/L引物、1U/25μL LA Taq酶(Takara)、2.5μL10×LATaq酶缓冲液,以及30ng基因组DNA,灭菌双蒸水补足体系。反应程序:95℃下5min,94℃下45s,依不同引物48~55℃退火1min,72℃延伸130s,72℃下5min,运行35个循环。PCR产物由1%的琼脂糖电泳检测,用宝生物纯化试剂盒回收PCR产物,纯化样品送至英俊测序公司进行测序。
1.3 序列分析
测序结果用软件CodonCode Aligner 2.0.1和Bioedit 7.0.1[16]进行拼接并辅以手工校对,获得完整线粒体控制区全序列。利用软件ClustalX1.8.3[17]进行序列比对排序,并辅以手工校对。使用MEGA 4.0[18]计算遗传距离和碱基组成,DAMBE4.5[19]程序计算出转换、颠换数与F84遗传距离,并做成散点图,来检验鳎类控制区比对数据核苷酸替代是否饱和。利用DNASIS v2.09搜索序列的发卡结构,并利用RNAFOLD[20]软件包,在最小自由能的原则下绘制出二级结构图。重复序列利用Tandem Repeats Finder[21]在默认参数下从控制区全序列中筛选。
2 结果
2.1 3种舌鳎亚科鱼类mtDNA控制区的基本特征
结果显示双线舌鳎、长钩须鳎和短钩须鳎的线粒体控制区全序列长度具有明显的差异,分别为655、1155和886bp,3种舌鳎鱼类的线粒体控制区与半滑舌鳎一样存在位移现象,由tRNAPro和tRNAPhe基因之间移位到了ND1基因下游;并且在5′末端都存在一段长度不等的重复序列。去掉重复序列获得其片段长度分别为620、741和704bp,差异依然明显。利用MEGA4.0计算CR序列的碱基含量,舌鳎亚科鱼类A、T、C、G含量分别为31.6%~35.9%、29.7%~34.7%、18.1%~22.0%、11.1%~15.6%,AT含量比较高(62.8%~68.6%),平均AT含量为66.3%,鲽形目其他四科代表鱼类的AT含量为(57.7%~63.3%),平均为60.4%(见表2)。
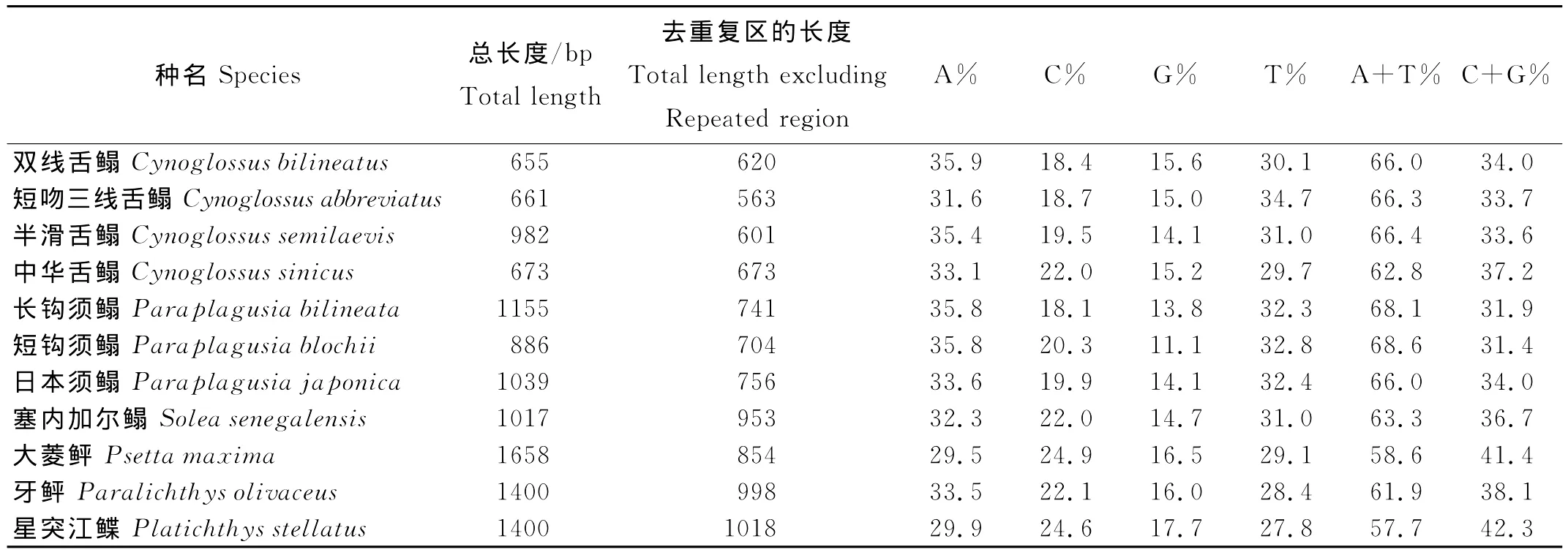
表2 11种鲽形目鱼类控制区长度和碱基组成信息Table 2 The information of the length and base composition of CRs from 11flatfish species

图1 7种舌鳎和2种鳎的控制区序列比对图。Fig.1 The sequences alignment of CRs from seven tonguefishes and two soles
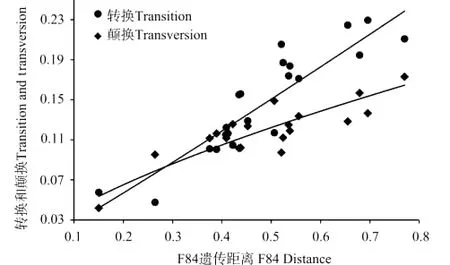
图2 7种舌鳎线粒体控制区比对序列的转换、颠换数与F84遗传距离的比对散点图Fig.2 Number of transition and transversion substitution versus F84distance in pairwise comparison for aligned CRs sequences of seven tonguefishes
将这3种舌鳎与GenBank中已有的舌鳎亚科的其他鱼类控制区序列比较发现,只有196bp长度的序列有相似度,其它位置则无法比对整齐;与鳎科的带纹条鳎和塞内加尔鳎比较则仅有22bp的序列有相似性(见图1)。转换、颠换的绝对数与遗传距离比较的散点图显示舌鳎线粒体控制区比对序列并没有达到饱和(见图2),所以其数据可以用来计算遗传距离及构建系统发育树。本研究中其它鲽形目鱼类则由于种类太少无法进行饱和性分析,但是随后计算其之间的遗传距离要小于舌鳎类,所以也可以证明其数据并未饱和,可以用来进行遗传距离分析。利用MEGA 4.0计算Kimura双参数遗传距离如表3,须鳎属内的遗传距离的范围从0.32~0.41,平均为0.35;舌鳎属内从0.11~0.28,平均为0.23;舌鳎属和须鳎属间从0.24~0.55,平均为0.40;舌鳎亚科内从0.11~0.55,平均为为0.34。舌鳎亚科鱼类控制区序列和其它鲽形目鱼类无法比对整齐,故无法计算遗传距离。鳎科的塞内加尔鳎、菱鲆科的大菱鲆、牙鲆科的牙鲆和鲽科的星突江鲽间的遗传距离从0.08~0.43,平均为0.26(见表3)。

表3 Kimura双参数遗传距离(对角线以上)和成比例的遗传距离(对角线以下)Table 3 Kimura 2-parameter genetic distances(above diagonal)and P-distances(below diagonal)
2.2 控制区序列的功能保守区域和重复序列特征
舌鳎亚科鱼类和其它鲽形目鱼类控制区的相似性极低,只有如图1所示的22bp左右的碱基对有相似性(图中标示为:鳎亚目保守序列区),这22bp保守序列块的序列通式为TCCAG-G-G-AAGGGG和赫崇波等[22]所提出的鲽形目CSB-A的序列通式AGCGAAGGGGTTCTCTTT的前半部分相似性高,而CSBB到CSB-F,及CSB 1-3等的保守模块则没有被识别。而其他4科鲽形目鱼类以4个代表种(塞内加尔鳎、大菱鲆、牙鲆和星突江鲽)进行比对后,有540bp左右长度的相似序列,赫崇波等所提出CSB A-F及CSB 1-3都能够从中被识别。
3种舌鳎科鱼类在5'末端都存在重复序列,拷贝次数和重复区长度都差异巨大,最长的重复区为长钩须鳎414bp,最短的双线舌鳎只有35bp(见表4)。3种舌鳎重复单元相似性较高,都含有保守序列块TACAT—ATGTA。通过DNASIS v2.09搜索发卡结构发现,3种舌鳎的重复区都可以形成稳定的发卡结构,短钩须鳎形成的发卡结构的茎区长14bp,最小自由能为-13.7kcal/mol,长钩须鳎形成的发卡结构最小自由能为-10.1kcal/mol(见图3),双线舌鳎虽然可以形成稳定的发卡结构,但是却没有发现如同长钩须鳎和短钩须鳎一样的长茎结构。
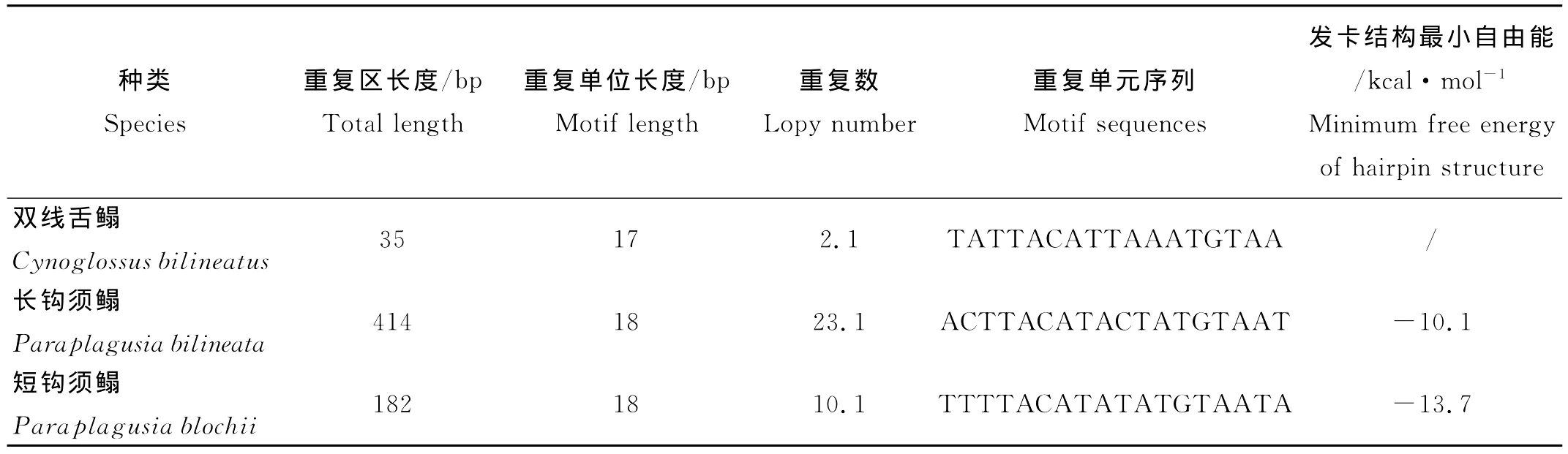
表4 3种舌鳎重复序列相关信息Table 4 The information of tandem repeats of CRs form three tonguefishes

图3 2种舌鳎重复序列形成的二级结构Fig.3 Secondary structures formed by repeated sequences in two tonguefishes
3 讨论
3.1 舌鳎类控制区的高速变异
3种舌鳎的线粒体控制区长度变化显著,长度655~1155bp,去掉重复区,依然存在较大的异质性。舌鳎亚科鱼类线粒体控制区相对于其它科的鲽形目鱼类变异极其显著,和鲽形目其它科的鱼类的控制区基本没有相似性(如图1所示只有二十几个碱基的保守序列),据此可以证明舌鳎亚科鱼类控制区的特殊性。
由于序列无法比对,所以不能进行舌鳎亚科鱼类和鲽形目其它鱼类的系统发育分析,因此本文只能通过比较舌鳎亚科鱼类及鲽形目其它科鱼类控制区序列相关的遗传距离来讨论舌鳎亚科鱼类控制区的异常变异。通过计算得到的舌鳎亚科内的Kimura双参数遗传距离范围从0.11~0.55,平均为为0.34。远大于以鳎科的塞内加尔鳎、菱鲆科的大菱鲆、牙鲆科的牙鲆和鲽科的星突江鲽为代表计算的科间的遗传距离(0.08~0.43,平均为0.26)。即便是须鳎属内(0.32~0.41,平均为0.35)的遗传距离也高于上述的非舌鳎类的种间遗传距离,虽然遗传距离并不能完全反映出物种的系统关系,但存在一定的参考价值,据此可以看出,舌鳎类的控制区变异十分异常,其变异速度远大于其它鲽形目鱼类,并且这种现象普遍存在于舌鳎亚科。
从碱基含量上来看,舌鳎亚科鱼类的AT含量也普遍高于非舌鳎鲽形目鱼类(见表2),舌鳎亚科鱼类AT含量从62.8%到68.6%,平均为66.3%,而其它4种鲽形目鱼类AT的平均含量仅60.4%。根据DNA碱基配对原则,A与T配对形成2个氢键,G与C相配形成3个氢键,所以GC之间的连接较为稳定。A+T的含量可以反映出序列的变异性,具有高比例的A+T可能是控制区序列变异较快的原因之一[23]。所以,从碱基组成中也能看出,舌鳎亚科鱼类控制区序列变异性较高。
将舌鳎亚科鱼类和鲽形目其它鱼类比较也仅仅发现类似CSB-A和TAS保守序列,CSB1-3和CSB B-F等区域都无法利用鲽形目的通式找到,而其他4种鲽形目鱼类(塞内加尔鳎、大菱鲆、牙鲆和星突江鲽)控制区序列则可以识别出所有这些保守序列,相互之间的相似度也高。这种存在于舌鳎亚科控制区的高速变异在硬骨鱼中尚未被报道。并且,硬骨鱼类1个亚科内如此多的种发生快速的变异也未见报道。
作者推测舌鳎亚科线粒体控制区这种快速的变异的原因有2种可能,1种是现存在舌鳎类的控制区是由原始的鲽类正常的控制区移位而来,伴随这种移位发生控制区的某些功能丧失,从而导致控制区稳定性减弱,稳定性减弱便是变异快速积累的直接原因。另1种是原来的控制区丢失,线粒体其他位置又进化成为新的控制区。目前还无法明确这2种可能哪一种是发生在舌鳎类控制区的真实事件,只有测定出更多的中间过程的控制区序列才有可能被揭示。
3.2 重复序列的特征及延伸过程的推测
3种舌鳎重复区单元都含有保守序列块TACAT—ATGTA(见表4)。这种保守块是由2段5 bp的反向互补序列组成,也就是说可以形成发卡结构,通过DNASIS v2.09搜索发卡结构发现其中3种舌鳎的重复区都可以形成非常稳定的长发卡结构。
Guo等[24]在鲤科鱼类识别了TAS保守序列为:TACATAT-ATGTATTATCACCATTATATTA
ACCA,并指出TAS-cTAS组件序列为TACAT-ATGTA。Kong等[6]和张艳春等[25]在研究鲽形目相关鱼类时同样识别了这一组件,舌鳎亚科重复序列的保守序列块和Guo等提出的TAS-cTAS组件序列完全相同,因此可以在重复序列中识别TAS序列。由于重复单元序列和Buroker等[13]描述的美洲鲟的重复单元类似,都存在TAS相关序列,并且同样位于控制区的5'末端,所以本文的重复区为Buroker的非正常延伸模型提供了新的证据。Buroker等[13]认为线粒体复制进行到重复区时,由于每个重复单元都存在TAS相关序列,所以线粒体的复制可以被终止在任何1个TAS上,之后被置换的重链和新生的线粒体重链竞争结合模板链,导致新生链和模板链融解,由于新生链可以折叠成稳定的二级结构,导致线粒体继续复制时新生链和模板链的结合容易发生错配,最终引起重复序列重复单元的增加或者减少。
作者利用非正常延伸模型结合舌鳎亚科重复区的数据模拟了舌鳎类重复单元延伸的过程。如图4所示。当线粒体复制进行到重复区的时候,由于多个重复单元都存在终止序列,所以线粒体的复制可以被终止在任何1个重复单元上(见图4A),当线粒体复制被终止后,由于被置换链的H链会和新生链竞争模板链会导致新生链的解链(见图4B),新生链单链暴露后,由于反向互补序列的存在便能形成稳定的二级结构(见图3),这种二级结构大大增加了新生链3'末端与模板链错配的几率(见图4C)。当线粒体的复制重现开始后就会导致重复单元增加(见图4D)。
尽管这些重复区形成的二级结构可以作为非正常延伸模型的一个证据,但是很多重复序列并不能形成这种二级结构,也不存在TAS相关序列,所以非正常延长模型并不是重复序列延伸的唯一的模型[26],要最终揭示重复区生成的真实过程,需要有更多的数据和进一步的研究。
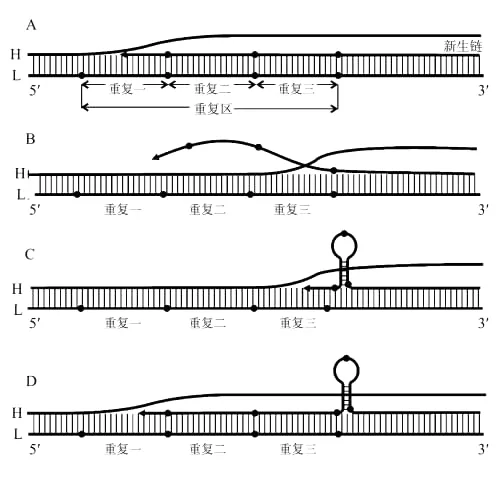
图4 根据非正常延伸模型推测的舌鳎重复区的延伸过程Fig.4 The speculated process of the elongation of repeated region in tonguefishes based on the illegitimate elongation model
[1] Nelson J S.Fishes of the world,4th edn[M].[s.l.]:John Wiley Inc,2006.
[2] 李思忠.中国动物志硬骨鱼纲鲽形目[M].北京:科学出版社,1995.
[3] Saitoh K,Hayashizaki K,Yokoyama Y,et al.Complete nucleotide sequence of Japanese flounder(Paralichthys olivaceus)mitochondrial genome:structural properties and cue for resolving teleostean relationships[J].J Hered,2000,91(4):271-278.
[4] Mcmillan W O,Palumbi S R.Rapid rate of control-region evolution in Pacific butterflyfishes(Chaetodontidae)[J].J Mol Evol,1997,45(5):473-484.
[5] Guo X,Liu S,Liu Y.Comparative analysis of the mitochondrial DNA control region in cyprinids with different ploidy level[J].Aquaculture,2003,224(1-4):25-38.
[6] Kong X Y,Yu J Z,Zhou L S,et al.Comparative analysis of 5'-end sequence of the mitochondrial control region of six flatfish species(Pleuronectidae)from the Yellow Sea[J].Raffles Bull Zool,2007:111-120.
[7] Broughton R E,Dowling T E.Evolutionary dynamics of tandem repeats in the mitochondrial DNA control region of the minnow Cyprinella spiloptera[J].Mol Biol Evol,1997,14(12):1187-1196.
[8] Lunt D,Whipple L,Hyman B.Mitochondrial DNA variable number tandem repeats(VNTRs):utility and problems in molecular ecology[J].Mol Ecol,1998,7:1441-1455.
[9] Tsaousis A D,Martin D P,Ladoukakis E D,et al.Widespread recombination in published animal mtDNA sequences[J].Mol Biol Evol,2005,22(4):925-933.
[10] Smith J M,Smith N H.Recombination in animal mitochondrial DNA[J].Mol Biol Evol,2002,19(12):2330-2332.
[11] Kraytsberg Y,Schwartz M,Brown T A,et al.Recombination of human mitochondrial DNA[J].Science,2004,304(5673):981-981.
[12] Levinson G,Gutman G A.Slipped-strand mispairing:a major mechanism for DNA sequence evolution[J].Mol Biol Evol,1987,4(3):203-221.
[13] Buroker N E,Brown J R,Gilbert T A,et al.Length heteroplasmy of sturgeon mitochondrial DNA:an illegitimate elongation model[J].Genetics,1990,124(1):157-163.
[14] Kong X,Dong X,Zhang Y,et al.A novel rearrangement in the mitochondrial genome of tongue sole,Cynoglossus semilaevis:control region translocation and a tRNA gene inversion[J].Genome,2009,52(12):975-984.
[15] Manchado M,Catanese G,Ponce M,et al.The complete mitochondrial genome of the Senegal sole,Solea senegalensis Kaup.Comparative analysis of tandem repeats in the control region among soles[J].DNA Seq,2007,18(3):169-175.
[16] Hall T A.BioEdit:a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT[J].Nucleic Acids Symposium Series,1999,41:95-98.
[17] Thompson J D,Gibson T J,Plewniak F,et al.The CLUSTAL_X windows interface:flexible strategies for multiple sequence alignment aided by quality analysis tools[J].Nucleic Acids Res,1997,25(24):4876-4882.
[18] Tamura K,Dudley J,Nei M,et al.MEGA4:Molecular Evolutionary Genetics Analysis(MEGA)software version 4.0[J].Mol Biol Evol,2007,24(8):1596-1599.
[19] Xia X,Xie Z.DAMBE:software package for data analysis in molecular biology and evolution[J].J Hered,2001,92(4):371-373.
[20] Zuker M,Stiegler P.Description of the method used in RNAFOLD[J].Nucleic Acids Res,1981,9:133-148.
[21] Benson G.Tandem repeats finder:aprogram to analyze DNA sequences[J].Nucleic Acids Res,1999,27(2):573-580.
[22] 赫崇波,曹洁,刘卫东,等.圆斑星鲽及相关种类线粒体DNA控制区结构分析[J].遗传,2007,29(7):829-836.
[23] 诸廷俊,杨金权,唐文乔.长江口鲚属鱼类线粒体DNA控制区结构分析[J].上海水产大学学报,2008,17(002):152-157.
[24] Guo X H,Liu S J,Liu Y.Comparative analysis of the mitochondrial DNA control region in cyprinids with different ploidy level[J].Aquaculture,2003,224(1-4):25-38.
[25] 张艳春,孔晓瑜,王忠明.大口鳒线粒体DNA控制区结构和鲽形目鱼类的系统进化初步研究[J].热带海洋学报,2010,29(6):71-78.
[26] Ray D A,Densmore L D.Repetitive sequences in the crocodilian mitochondrial control region:Poly-A sequences and heteroplasmic tandem repeats[J].Mol Biol Evol,2003,20(6):1006-1013.
Preliminary Study on the Rapid Evolution of MtDNA Control Region and the Elongated Mechanism of Tandem Repeat Units in Cynoglossinae Fishes
SHI Wei1,2,KONG Xiao-Yu2,JIANG Jin-Xia2,MIAO Xian-Guang2
(1.College of Fisheries,Ocean University of China,Qingdao 266003,China;2.Marine Biodiversity Collection of South China Sea,South China Sea Oceanology Institute of Chinese Academy of Sciences,Guangzhou 510301,China)
The complete control regions of mitochondrial DNA were amplified in three tongue soles Cynoglossus bilineatus(Lacepède,1802),Paraplagusia bilineata(Bloch,1787),and Paraplagusia blochii(Bleeker,1851)from subfamily Cynoglossinae,and compared with that from other four flatfishes.The length of control regions(CR)ranged from 655to 1155bp and exhibited apparent length heteroplasmy.Comparisons showed that the control regions have significant differences from that of other flatfishes,with only conserved blocks of CSB-A and TAS identified and higher AT content detected.It was presumed that these variations may have resulted in the fast evolution of CR regions in these tongue soles.Furthermore,the presence of tandem repeats at 5'end of control regions was detected,and these repeat units bear TAS-like sequences,which the possess capability of forming the stable secondary structure in all three tongue soles.These characteristics of control regions in those soles provide likely new evidences for the illegitimate elongation model,the elongated mechanism of tandem repeat units.Additionally,the elongation process of these tandem repeats in tongue soles was speculated using illegitimate elongation model.
Pleuronectiformes;Cynoglossinae;control regions;tandem repeat;illegitimate elongation
Q349;Q959.486
A
1672-5174(2012)1-2-081-07
国家自然科学基金项目(30870283;31071890)资助
2011-04-29;
2011-05-04
时 伟(1984-),男,博士。E-mail:kcool@126.com
**通讯作者:E-mail:xykong@scsio.ac.cn
责任编辑 王 莉
猜你喜欢
遗传(2022年9期)2022-10-10
海洋信息技术与应用(2022年1期)2022-06-05
海洋通报(2020年5期)2021-01-14
儿童时代·幸福宝宝(2020年9期)2020-09-08
现代企业文化·理论版(2020年16期)2020-08-31
武夷科学(2019年2期)2019-12-20
中国公路(2017年18期)2018-01-23
