
凡纳滨对虾肠道微生物宏基因组Solexa测序及其初步分析
2012-10-23 03:01严雪平袁剑波
海洋科学 2012年6期
严雪平, 袁剑波, 刘 斌
(1. 中国科学院 海洋研究所 实验海洋生物学重点实验室, 山东 青岛 266071; 2. 中国科学院 研究生院, 北京 100049)
凡纳滨对虾肠道微生物宏基因组Solexa测序及其初步分析
严雪平1,2, 袁剑波1,2, 刘 斌1
(1. 中国科学院 海洋研究所 实验海洋生物学重点实验室, 山东 青岛 266071; 2. 中国科学院 研究生院, 北京 100049)
CTAB-酚/氯仿法提取新鲜凡纳滨对虾(Litopenaeus vannamei)肠道微生物宏基因组DNA。结果表明: CTAB-酚/氯仿法提取总DNA的浓度达到92.5 ng/μL, 半定量PCR法检测微生物基因组相对含量为69.9%。凡纳滨对虾肠道微生物宏基因组用于 Solexa测序, 生物信息学分析结果显示宏基因数据中64.1%的数据属于未知序列, 35.5%属于真核生物, 而已知的微生物和病毒序列所占比例仅有0.4%。
肠道微生物宏基因组; CTAB; Solexa测序
海洋动物肠道内共附生着大量的微生物, 形成了一个特定的微生态群落。研究海洋动物肠道微生物的基因组结构有利于理解这类微生物的生理生化特征, 对开发海洋微生物产生的抗肿瘤、抗菌等生物活性物质有着重要的意义[1]。但因现有培养技术的局限性, 从可培养微生物中获得活性物质越来越难[2]。自1998年Handelsman等[3-4]提出宏基因组学的概念以来, 宏基因组学在微生物资源的开发、活性物质的筛选等方面等得到了广泛的应用但是宏基因组测序费用昂贵、所需基因组DNA至少在10 μg以上, 而实际上有些宏基因组 DNA难以获得, 且常有外源DNA的污染, 因此获得高纯度足量的宏基因组DNA成为宏基因组研究瓶颈之一[5]。在现实工作中往往需要大量采样、大量提取基因组才能满足要求, 不仅费时费力且容易造成实验误差和结果的不稳定, 又由于宏基因组测序往往是大规模、高通量, 费用昂贵、测得序列数据量大, 序列拼接组装繁杂耗时, 因此宏基因组测序前进行 DNA的定性定量检测非常必要。半定量PCR技术经常用于评估样本中靶基因的分子数, 实现对核酸信息的量化分析及比较, 结果可靠、操作方便[6-10]。但目前, 有关这方面的报道还不多。研究探索一种微生物宏基因组DNA评价方法在宏基因组研究方面具有重要的意义。
水生动物与陆生恒温动物一样肠道微生物丰富,其中有相当一部分微生物长期定居在动物肠道中,与宿主的营养、代谢、免疫等一些列生理生化过程密切相关[11-13]。研究人员发现在人及多种动物中, 多种生理功能是有定居在肠道中的微生物的参与得以实现[14-15], 例如: 2010年Hehemann[16]研究发现存在于海洋杆菌(Zobellia galactanivorans)基因组中编码Porphyranases酶的基因也存在于人肠道微生物日本人肠道杆菌(Bacteroides plebeius)基因组中, 这种酶能帮助人分解利用海藻植物类营养, 肠道宏基因组的比较分析表明 Porphyranases在日本人中常见, 但并不存在于北美人群中。凡纳滨对虾(Litopenaeus vannamei)作为重要的海、 淡水经济养殖对虾, 凡纳滨对虾是寻找益生菌和抗菌、抗肿瘤活性物质的重要物种之一[12,17]。作者以凡纳滨对虾肠道微生物为研究对象, 提取了肠道微生物宏基因组 DNA, 以细菌16S为靶基因、凡纳滨对虾18S rDNA为内参对微生物宏基因组中宿主DNA污染的程度进行半定量研究, 以期建立一种便捷、可靠的检测宏基因组 DNA纯度和浓度的方法。同时对测得宏基因组数据进行初步生物信息学分析, 进行相关物种鉴定分析和宏基因组功能初步分析。
1 材料与方法
1.1 材料、主要试剂
健康成体凡纳滨对虾(体长 10~14 cm)采自青岛东海湾对虾养殖场。微生物基因组提取试剂盒(DP302-02)购于天根生物科技有限公司。
1.2 方法
1.2.1 宏基因组DNA提取
青岛东海湾对虾养殖场采健康成体凡纳滨对虾45尾, 实验室条件下养殖 48 h, 以尽量排空肠道。70%酒精体表消毒, 无菌操作取出整个肠道。肠道浸泡于PBS缓冲, 充分振荡5~10 min后1000 r/min离心 10 min, 取上清。PBS洗涤 3次肠道, 合并上清10 000 r/min离心10 min收集菌体。PBS清洗3次。收集到的菌体以 CTAB-酚/氯仿法提取基因组。CTAB-酚/氯仿法参考李可[17]、Fridez等[18]。最后以200 μL TE 溶解 DNA。
1.2.2 宏基因组DNA定量
提取的宏基因组DNA通过1%琼脂糖凝胶电泳检测, 同时利用微量紫外可见分光光度计 NanodropND-1000分别测定其浓度和纯度, 重复3次。
1.2.3 半定量PCR法评价宏基因组DNA
以细菌 16S rDNA为靶基因, 凡纳滨对虾 18S rDNA为内参, 分别代表细菌和宿主基因组, 进行半定量PCR检测所提取宏基因组中微生物基因组和宿主凡纳滨对虾基因的相对含量。设置3组平行对照,重复 3次, 每组取两个 PCR反应结果电泳检测。Bio-Rad的1D凝胶定量生物学软件Quantity One -4.6.2分析电泳检测结果。
1.2.3.1 引物设计
根据 GenBank(http://www.ncbi.nlm.nih.gov)上的凡纳滨对虾的 18S序列设计引物: F3445: 5’-TAGGGGTGTTGGGGACG-3’, B4795: 5’-AACATTGTCTTTCCCACGC-3’, 片段长度为 1034bp。细菌 16S通用引物, 27-F: 5’-AGAGTTTGATCCTGGCTCAG-3’,1492-R: 5’-CGCCAGGGTTTTCCCAGTCACGAC-3’,扩增片度长度约为 1 500 bp。引物由生工生物工程(上海)有限公司合成。
1.2.3.2 半定量PCR的条件
以提取的宏基因组为模板进行半定量 PCR检测。PCR反应按照25 μL体系。半定量PCR的反应条件: 95℃5 min, 94℃45 s, 57.9℃50 s, 72℃1 min10 s, 26 循环, 72℃10 min。
1.3 凡纳滨对虾肠道微生物宏基因组Solexa测序及初步分析
将提取的凡纳滨对虾肠道微生物宏基因组DNA送杭州华大基因用于Solexa测序, Perl编程统计出所有reads的每个位点的4种碱基频率和质量分数值去除测序质量较差的reads, Velvet软件拼接成contigs,进一步采用 Blast 将虾的宏基因组序列组装文件Shrimp.contigs 利 用 NCBI(http://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastn&BLAST_PROGRA MS=megaBlast&PAGE_TYPE=BlastSearch&SHOW_DEFAULTS=on&LINK_LOC=blasthome)上 Nt 数据库进行比对(e 值限定为 1e-5), 得到 Blast 的文件,生物信息学分析参考Mitra等[19-22]。
2 结果与分析
2.1 宏基因组DNA电泳检测及紫外分光光度计定量检测
从电泳结果看, CTAB-酚/氯仿法提的总 DNA片段均比较完整, 条带清晰明亮, 主带在 23kbp(图1)。紫外吸光光度计测得数据表明所提取的宏基因紫外吸收峰在260 nm处, OD260/OD280的比值平均值为1.75, 表明所提取的宏基因组 DNA质量较好。浓度检测结果显示所提取的宏基因组 DNA浓度为 92.5 mg/L, 最终所提取的总DNA量为18.5 μg。
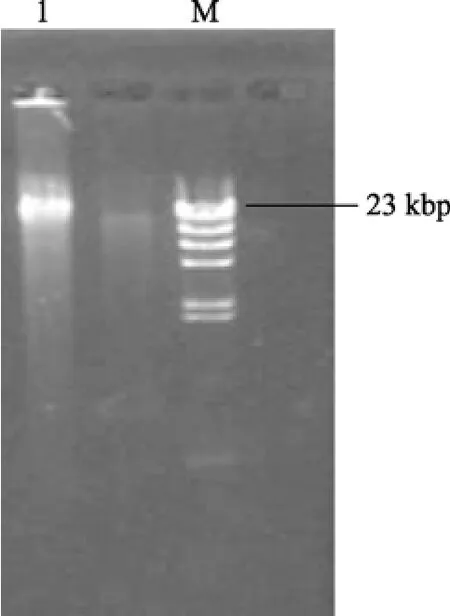
图1 提取肠道微生物宏基因组电泳检测Fig. 1 The gel electrophoresis of intestinal microflora metagenomeM.λ-Hind III digest maker; 1.CTAB-酚/氯仿法提取的样品宏基因组DNAM.λ-Hind III digest maker; 1. Metagenome DNA extracted by CTAB- phenol/chloroform protocol
2.2 宏基因组DNA半定量PCR检测
Quantity One分析电泳检测结果表明: CTAB-酚氯仿提取新鲜样品中微生物基因组平均相对含量为69.9%。
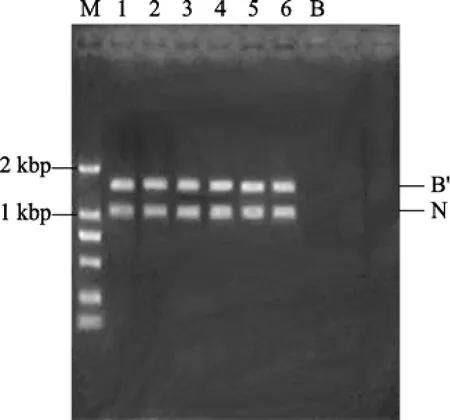
图2 半定量测定提取宏基因组中微生物基因组的含量Fig. 2 The content of bacterial genomes in metagenome detected by Semi-quantitative PCRM. DL2000 marker; 1-6. 宏基因组半定量PCR扩增; B. blank对照; B’. 细菌16S; N. 凡纳滨对虾18SM. DL2000 marker; 1-6. Metagenome as template for semi-quantative PCR; B. Blank control; B’. Bacterial 16S DNA; N:Shrimp 18S DNA
2.3 宏基因组 DNA Solexa测序结果生物信息学初步分析
由于用于Solexa测序所需DNA至少在10 μg以上, 所提基因组达到测序要求。将所提宏基因组DNA用于新一代测序技术 Solexa测序法测序。Solexa测序总共得到4.65G的数据20441981条reads,共计3924860352个碱基, GC含量为49.65%, 进一步将所得到的reads序列经去除引物接头等冗余序列以Velvet软件拼接组装成contigs。在此基础上, 将拼接的contigs中重复的、序列过短、分值不高的contigs去除, 选取其中的一个最优匹配结果作为该 contigs的比对信息, 这样就得到的是每一条 contig对应唯一的一个比对信息得到19994条contigs。作者进一步将虾的宏基因组序列组装contigs序列与 Nt 数据库进行 Blast比对(e 值限定为 1e-5), 得到 blast 的结果用 MEGAN 软件分析相关物种的丰度和进化关系图结果如图 3 (圆圈越大, 代表比对上的 contig越多), 结果显示只有 31条contigs个是与细菌基因组序列相关(大部分为弧菌属Vibrio), 有 7 106条contigs与真核生物序列相关, 24条contigs为病毒序列。其余的contigs均是已知序列但是功能未知(1 856条)和未比对上 contigs(10 977条), 表明宏基因组测序中含有较多未知物种, 他们的序列都未被测定,所以需要进一步的实验验证。与数据中占35.5%的真核生物序列比对上的物种类群主要有: (1)凡纳滨对虾数据4 701条; (2)顶复动物亚门的物种(原生动物)相关序列1 290条。数据中占64.1%的未知功能序列和未比对上序列极有可能是虾肠道中独特的或未被测定的微生物种群基因组序列, 研究这些序列有助于进行相关未培养的细菌研究。
作者进一步将所得宏基因组数据用 MG-RAST软件(e值限定在1e-5, 采用的数据库是NCBI数据库,不限定比对长度)分析, 结果与 MEGAN软件分析较一致。序列功能分析采用的数据库是NOG和KEGG的数据库(e值限定在 1e-5, 不限定比对长度), 分析结果显示所得宏基因组数据中与细菌细胞分裂相关功能项包含序列最多, 此外 RNA代谢, 碳水化合物代谢还有氨基酸代谢和衍生物合成这 3大功能项包含的序列也较多。此外, 作者分别下载NCBI中所有质粒序列和抗性质粒序列, 构建成质粒数据库和具有抗性的质粒数据库, 将所测宏基因组数据与质粒数据库和抗性质粒数据库进行比对, 发现其中34条contigs与质粒数据相关, 具有抗性质粒相关的contigs 29条。其中抗性相关序列中有12条contigs与固氮螺菌(Azospirillumsp.)所含质粒序列相关, 11条 contigs与耐金属贪铜菌(Cupriavidus metallidurans)中的质粒序列相关。
3 讨论
随着宏基因组学的不断发展, 越来越多的环境微生物和动物肠道微生物正在被人们重视[6]。如何获得这些肠道固有微生物的基因组并有效地减少宿主的基因组和外源DNA是研究动物肠道微生物宏基因组的关键[2,5,17]。本实验中, 作者采用 CTAB-酚氯仿法提取新鲜样品组 DNA, 其基因组 DNA浓度达到92.5 mg/L, 且DNA片段完整。半定量PCR检测微生物相对含量为 69.9%, 与宏基因组测序结果比较存在一定的偏差, 可能是由于本实验只扩增了细菌16S, 忽略了古细菌和真核微生物(真菌、原生动物)等, 这会使微生物基因组的含量在结果中偏低。其次,选择单个基因作为靶基因、内参基因, 在一定程度上忽略了微生物基因组和凡纳滨对虾染色体基因组大小的差异, 容易造成微生物基因组结果偏高。因此,可设置多个内参以便更科学地测算宿主与微生物基因组相对比。宏基因组技术在研究环境及未可培养微生物活性物质方面有着广泛的应用[23-24], 本研究中宏基因测序结果分析表明已知微生物基因组序列比较较少, 但其中含有大量的序列属于已被其他研究测定但功能未知微生物基因组序列, 提示是虾肠道中含有较多的独特的或未被测定的微生物种群。所测序列中含有真核生物相关序列较多, 这可能与虾肠道中寄生有原生动物有关。宏基因组数据中存在大豆、玉米等高等植物基因组, 这可能与虾饵料等因素相关[25]。在宏基因组数据中, 发现部分的质粒相关序列, 显示虾肠道微生物中可能含有编码相关抗生素的某些基因。作者的研究为宏基因组DNA提取及检测提供了很好的思路, 宏基因组测序和生物信息学分析对这类特殊微生物群落的遗传特性、生理生化功能基本特征有初步了解, 为虾类病虫害的防治以及健康养殖等应用研究提供了一定科学依据。
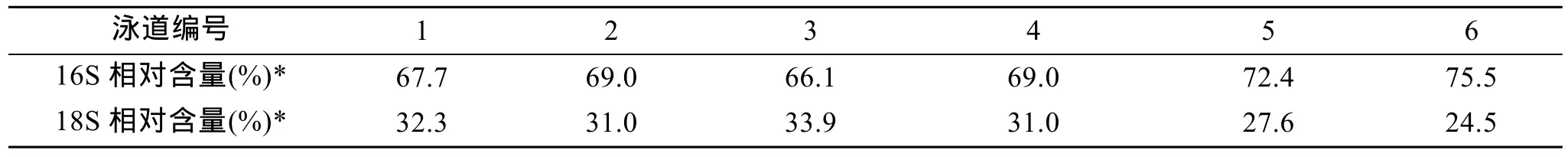
表1 Quantity One-4.6.2分析半定量PCR条带的相对含量Tab. 1 Quantity One-4.6.2 analysis of semi-quantitative PCR electrophoresis bands
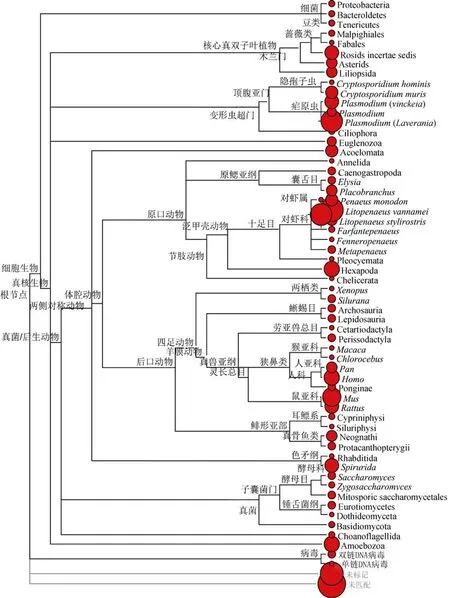
图3 MEGAN软件分析宏基因组数据相关物种丰度和进化关系树Fig. 3 MEGAN software analysis abundance and Phylogenetic tree of species in metagenome
[1] Glad T, Bernhardsen P, Nielsen K M, et al. Bacterial diversity in faeces from polar bear (Ursus maritimus)[J].Arctic Svalbard BMC Microbiol, 2010, 10(10): 1-10.
[2] 李慧, 何晶晶, 张颖, 等.宏基因组技术在开发未培养环境微生物基因资源中的应用[J]. 生态学报, 2008,28(4): 1762-1771.
[3] Handelsman J, Rondon M R, Brady S F, et al. Molecular biological access to the chemistry of unknown soil microbes: a new frontier for natural products[J]. Chem Biol, 1998, 5(10): 245-249.
[4] 李丽娟, 张殷昌, 龚世园. 宏基因组技术在开发未培养微生物资源中的应用[J]. 水利渔业, 2007, 27(3):7-9.
[5] Poniat H N, Schearz C, Qi J, et al. Metagenomics to paleogenomics: large-scale sequencing of mammoth DNA [J]. Sci, 2006, 311: 392-394.
[6] 孙奋勇, 万大方, 覃文新, 等. 基因组半定量PCR方法检测肿瘤组织的基因缺失[J]. 肿瘤, 2000, 20(2):135-137.
[7] 李春艳, 高文信, 张茹慧, 等.半定量 PCR 检测口腔鳞状细胞癌凋亡蛋白酶活化因子的表达[J] .实用口腔医学杂志, 2008, 24(6): 110-112.
[8] 刘青, 魏华. 两种半定量 PCR方法在鲫雌激素样效
应研究中的应用[J]. 水产学报, 2006, 30(6): 416-420.[9] Salone A, Nikkila J, Tuovinen J J, et al. Comparative analysis of fecal DNA extraction methods with phylogenetic microarray: effective recovery of bacteria and archaeal DNA using mechanical cell lysis[J]. J Microbiol Methods, 2010, 81(2): 127-134.
[10] 马媛媛, 邹少兰, 张鲲, 等. 半定量 RT-PCR 检测运动发酵单胞菌中外源基因转录水平的研究[J]. 微生物学通报, 2009, 36(6): 831-836.
[11] Navarrete P, Espejo R T, Romero J. Molecular analysis of microbiota along the digestive tract of juvenile Atlantic salmon (Salmo salar L.)[J]. Microb Ecol, 2009,57(3): 550-561.
[12] Wang X H, Li H R, Zhang X H, et al. Microbial flora in the digestive tract of adult penaeid shrimp(Penaeus chinensis)[J]. Journal of Ocean University of Qingdao,2000, 30(3): 493-498.
[13] Worthen P L, Gode C J, Graf J. Culture-independent characterization of the digestive-tract microbiota of the medicinal leech reveals a tripartite symbiosis[J]. Applied and Environmental Microbiology, 2006, 72(7):4775-4781.
[14] Tannock G W. Molecular assessment of intestinal microflora[J]. AJCN, 2001, 73 (suppl): 410-414.
[15] Kurokawa K, Itoh T, Kuwahata T, et al. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes [J]. DNA Res, 2007, 14(4):169-181.
[16] Hehemann H J, Correc G, Barbeyron T, et al. Transfer of carbohydrate-active enzymes frommarine bacteria to Japanese gut microbiota[J]. Nat, 2010, 464: 908-914.
[17] 李可, 郑天凌, 田蕴, 等. 南美白对虾肠道微生物群落的分子分析[J]. 微生物学报, 2007, 47(4): 649-653.
[18] Fridez F, Coquoz R. PCR DNA typing of stamps:evaluation of the DNA extraction[J]. Forensic Sci Int,1996, 78(2): 103-110.
[19] Mitra S, Klar B,Huson D H. Visual and statistical comparison of metagenomes[J]. Bioinformatics, 25(15):1849-1855.
[20] Mitra S, Gilbert J A, Field D, et al. Comparison of multiple metagenomes using phylogenetic networks based on ecological indices[J]. The ISME Journal, 2010,4: 1236-1242.
[21] Poinar H N, Schwarz C, Qi J, et al. Metagenomics to paleogenomics: large-scale sequencing of mammoth DNA[J]. Sci, 2006, 311: 392-394.
[22] Huson D H, Auch A F, Qi J, et al. MEGAN analysis of metagenomic data [J]. Genome Res, 2007, 17(3):377-386.
[23] Grant S, Grant W D, Cowan D, et al. Identification of eukaryotic open reading frames in metagenomics cDNA libraries made form environmental samples[J]. Aem,2006, 72(1): 135-143.
[24] Cowan D, Meyer Q, Stafford W, et al. Metagenomic gene discovery: past, present and future[J]. Trends Biotechnol, 2005, 23(6): 321-329.
[25] Oxley A P A, Shipton W, Owens L, et al. Bacterial flora from the gut of wild and cultured banana prawn,Penaeus merguiensis[J]. J Appl Microbiol, 2002, 92(2):214-223.
Solexa sequencing and analysis of the intestinal microorganisms metagenome in Litopenaeus vannamei
YAN Xue-ping1,2, YUAN Jian-bo1,2, LIU Bin1
(1. Key Laboratory of Experimential Marine Biology, Institute of Oceanology, Chinese Academy of Science, Qingdao 266071, China; 2.Graduate University of Chinese Academy of Science, Beijing 100049,China)
Apr., 15,2011
intestinal microfloar metagenome; CTAB; solexa sequencing
The intestinal microorganisms metagenome of Litopenaeus vannamei was extracted by CTAB-phenol/chloroform DNA extraction protocol. The relative concentration of bacterial genome in metagenome was analyzed by semi-quantitative PCR. The results showed that the total concentration of metagenome was 92.5 mg/L and relative microbial metagenome ratio was 69.9%. Solexa sequencing showed that up to 64.1% of contigs in metagenome had no assignment or no hits in database of NCBI, 35.5% of which were assigned to eukaryon, and only 0.4% sequences belonged to bacteria and virus.
Q89
A
1000-3096(2012)06-0009-06
2011-04-15;
2011-05-16
国家高技术研究发展计划(2007AA09Z444)
严雪平(1985-), 男, 硕士研究生, 主要从事海洋微生物宏基因组研究, 电话: 0532-82898957, E-mail: yanxueping007@126.com; 刘斌, 通信作者, 电话: 0532-82898857, E-mail: bliu@qdio.ac.cn
(本文编辑:谭雪静)
猜你喜欢
当代水产(2022年8期)2022-09-20
当代水产(2022年5期)2022-06-05
当代水产(2021年8期)2021-11-04
当代水产(2021年4期)2021-07-20
世界科学技术-中医药现代化(2020年2期)2020-07-25
当代水产(2019年1期)2019-05-16
中成药(2018年12期)2018-12-29
中成药(2017年6期)2017-06-13
广东海洋大学学报(2015年4期)2016-01-13
医学研究杂志(2015年4期)2015-06-10
