
嘧啶酮衍生物的微波合成*
2012-11-21 01:10刘永亮郭宝铭钟为慧
合成化学 2012年1期
马 旺, 刘永亮, 郭宝铭, 钟为慧
(浙江工业大学 药学院 浙江省制药工程重点实验室,浙江 杭州 310014)
嘧啶酮衍生物是一类重要的杂环化合物,具有抗免疫[1]、抗艾滋[2]、抗神经衰弱[3]、抗肿瘤[4]、抗菌[5]等药理活性。合成4H-嘧啶[1,2-b]哒嗪-4-酮的传统方法[6~8]由3-(二甲基氨基)丙烯酸与3-氨基哒嗪在乙酸中反应完成。
Baylis-Hillman反应是一类形成碳-碳键、生成多个官能团分子的有效方法,由于Baylis-Hillman加成物(1a~1k)具有多个能进一步转换的官能团,被广泛用于杂环化合物的合成[9~13]。
本文借鉴文献方法,设计了新的合成路线:在微波辅助下,1a~1k与4-氨基-6-氯嘧啶(2)或2-氨基-噻唑(3)反应,快速合成了两类嘧啶酮衍生物——3-取代-7-氯-4H-嘧啶[1,2-b]哒嗪-4-酮(4a~4k)和6-取代-5H-噻唑[3,2-a]嘧啶-5-酮(5a,5g,5i,5j, Scheme 1),收率81%~98%,其结构经1H NMR,13C NMR, IR和MS确证。
1 实验部分
1.1 仪器与试剂
B-540型电热熔点仪(温度计未校正);Varian-400 MHz型核磁共振仪(DMSO-d6为溶剂,TMS为内标);Thermo NicoletAvatar 370型红外光谱仪(KBr压片);Thermo Finnigan LCQ型质谱仪;APEX (Bruker) mass Ⅲ型质谱仪;APEX微波合成仪。所用试剂均为分析纯。

Scheme1
1.2 4的合成通法
氮气保护,在反应瓶中依次加入1a~1k2 mmol, 2 2 mmol的乙酸溶液10 mL,混合均匀后将反应瓶置微波反应器中,磁力搅拌下于115 ℃/300 W反应5 min~15 min。冷却至室温,加冰水(50 mL)析出沉淀,过滤,滤饼依次用水、乙醇洗涤,减压干燥得4a~4k。实验结果与表征数据分别见表1和表2。

表 1 合成4的实验结果Table 1 Experimental results of synthesizing 4

表 2 4的表征数据Table 2 Characteristic data of 4
续表2

Comp1H NMR δ(J/Hz)13C NMR δ(J/Hz)IR ν/cm-14c3.76(s, 2H, CH2), 7.25~7.28(m, 1H, ArH), 7.29~7.32(m, 2H, ArH), 7.41(t, J=0.4, 1H, ArH), 7.71(d, J=9.6, 1H, b-H), 7.74(d, J=9.6, 1H, a-H), 8.51(s, 1H, c-H)33.0, 126.2, 127.0, 127.6, 128.5, 130.0, 130.1, 132.8, 134.7, 139.1, 141.1, 145.6, 147.3, 167.01 642, 1 541, 1 4894d3.75(s, 2H, CH2), 7.32~7.38(m, 4H, ArH), 7.71(d, J=9.6, 1H, b-H), 7.74(d, J=9.6, 1H, a-H ), 8.45 (s, 1H, c-H)32.7, 127.4, 128.1(2C), 130.1, 130.7(2C), 130.9, 134.7, 137.6, 138.9, 145.6, 147.3, 167.03 089, 3 052, 1 625, 1 541, 1 4864e3.90(s, 2H, CH2), 7.26~7.31(m, 1H, ArH), 7.39~7.43(m, 2H, a,b-H), 7.62(s, 1H, ArH), 7.32(s, 1H, c-H)25.1(d, J=3.0), 114.7(d, J=22.8), 122.7(d, J=18.2), 125.5(d, J=25.8), 125.7, 129.9(d, J=9.9), 130.4, 134.7, 134.8, 137.4, 145.9, 147.3, 161.1(d, J= 245.7), 166.73 033, 1 633, 1 594, 1 4784f3.90(s, 2H, CH2), 7.59(t, J=8.0, 1H, ArH), 7.71(d, J=9.6, 1H, b-H), 7.76(d, J=9.6, 1H, a-H), 7.84(d, J=8.0, 1H, ArH), 8.08(dq, J=1.2, 8.0, 1H, ArH), 8.24(t, J=1.2, 1H, ArH), 8.63(s, 1H, c-H)33.0, 121.4, 123.4, 126.6, 129.6, 130.2, 134.8, 135.8, 139.4, 140.9, 145.7, 147.4, 147.7, 167.03 084, 1 644, 1 541, 1 3444g3.72(s, 5H, CH2, OCH3), 6.78(dd, J=2.8, 8.0, 1H, ArH), 6.89(s, 1H, ArH), 6.91(d, J=2.8, 1H, ArH), 7.20(t, J=8.0, 1H, ArH), 7.70(d, J=9.6, 1H, b-H), 7.74(d, J= 9.6, 1H, a-H), 8.32(s, 1H, c-H)33.3, 54.9, 111.6, 114.7, 121.1, 127.8, 129.2, 130.1, 134.7, 138.7, 139.9, 145.5, 147.1, 159.2, 167.03 095, 1 643, 1 581, 1 4894h3.74(s, 2H, CH2), 7.11(t, J=8.8, 2H, ArH), 7.38(dd, J=5.6, 8.8, 2H, ArH), 7.70(d, J=9.6, 1H, b-H), 7.74(d, J=9.6, 1H, a-H), 8.41(s, 1H, c-H)32.6, 114.9(J=21.2, 2C), 127.8, 130.2, 130.7(J=8.3, 2C), 134.6, 134.7, 138.8, 145.6, 147.2, 160.9(J=240.4), 167.13 087, 3 008, 1 642, 1 543, 1 4934i3.96(s, 2H, CH2), 6.94~6.98(m, 2H, ArH), 7.34(dd, J=0.8, 4.8, 1H, ArH), 7.71(d, J=9.6, 1H, b-H), 7.75(d, J=9.6, 1H, a-H), 8.39(s, 1H, c-H)27.6, 124.6, 126.1, 126.8, 127.3, 130.2, 134.7, 138.7, 140.3, 145.7, 147.2, 166.83 082, 1 641, 1 541, 1 4904j0.90(s, 3H, CH3), 0.92(s, 3H, CH3), 1.40~1.46(m, 2H, e-H), 1.52~1.59(m, 1H, d-H), 2.42(t, J=7.2, 2H, f-H), 7.70(d, J=9.6, 1H, b-H), 7.74(d, J=9.6, 1H, a-H), 8.37(s, 1H, c-H)22.3(2C), 25.7, 27.3, 36.1, 129.1, 130.0, 134.7, 137.9, 145.4, 146.9, 167.43 033, 2 951, 1 634, 1 593, 1 541, 1 4824k0.91(t, J=6.8, 3H, CH3), 1.26~1.39(m, 4H, e-H), 1.62~1.67(m, 2H, d-H), 2.58(t, J=7.6, 2H, f-H), 7.34(d, J=10.0, 1H, b-H), 7.60(d, J=10.0, 1H, a-H), 7.92(s, 1H, c-H)13.9, 21.9, 26.6, 27.7, 31.0, 128.9, 130.0, 134.7, 137.9, 145.4, 146.9, 167.43 034, 2 925, 1 635, 1 593, 1 541, 1 481
2 结果与讨论
2.1 实验条件优化
以1a作底物合成4a为例,无溶剂或乙酸作溶剂,对比传统加热方式与微波合成,结果见表3。由表3可见,微波合成更清洁、经济,收率最高可达95%。

表 3 4a的微波合成与常规合成法的比较Table 3 Synthesis comparison between microwave irradiation and conventional heating of 4a
2.2 底物结构对反应的影响
以乙酸为溶剂,于115 ℃反应5 min,其余反应条件同1.2,比较底物结构对反应的影响,结果见表4。从表4可以看出,当1a和1b的酯基由-OMe变为-OEt时,4a和4b的收率都略有下降;当1的R由芳基变为烷基(1j和1k)时,4j和4k的收率也明显降低。

表 4 底物结构对反应的影响Table 4 Effect of substrate on the yield of 4
2.3 扩展反应
以乙酸为溶剂,1a,1g,1i或1j与3于115 ℃反应5 min,其余反应条件同1.2制得5a,5g,5i和5j,其结果与文献[14]方法的比较见表5。由表5可见,微波合成不仅大缩短了反应时间,而且可以提高收率。
5a: 淡黄色晶体,m.p.227.9 ℃~230.1 ℃;1H NMRδ: 3.87(s, 2H, CH2), 7.21~7.98(m, 6H, ArH), 7.73(d,J=4.4 Hz, 1H, g-H), 8.16(s, 1H, c-H); ESI-MSm/z: 243(M++1, 100%)。
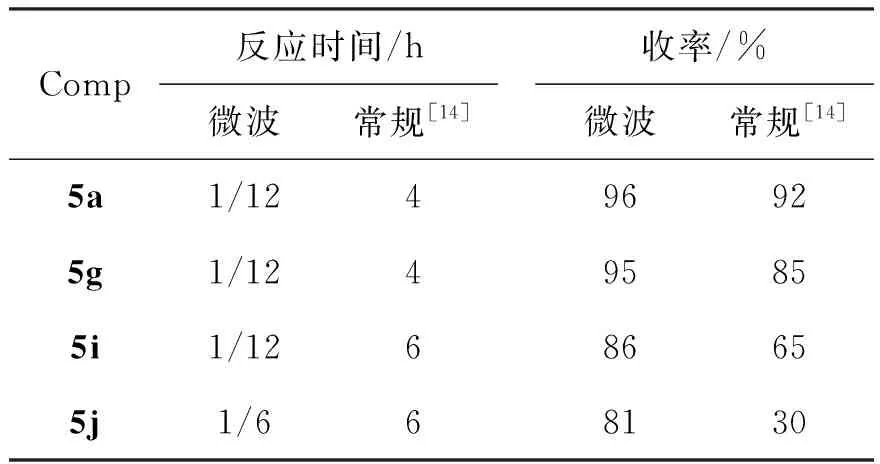
表 5 5的微波合成*与常规合成法的比较Table 5 Synthesis comparison between microwave irradiation and conventional heating of 5
*以乙酸为溶剂,1a,1g,1i或1j与3于115 ℃反应5 min,其余反应条件同1.2
5g: 灰色粉末,m.p.204.1 ℃~205.1 ℃;1H NMRδ: 3.65(s, 2H, CH2), 3.73(s, 3H, OCH3), 6.79(dd,J=2.8 Hz, 8.4 Hz, 1H, ArH), 6.82(d,J=9.2 Hz, A1H, rH), 6.87(s, 1H, ArH), 7.21(t,J=7.2 Hz, 1H, ArH), 7.26(d, 1H,J=4.4 Hz, h-H), 7.73(d,J=4.4 Hz, 1H, g-H), 8.13(s, 1H, c-H); ESI-MSm/z: 273(M++1, 100%)。
5i: 灰色粉末,m.p.192.5 ℃~193.2 ℃;1H NMRδ: 3.89(s, 2H, CH2), 6.96(m, 2H, h-H, ArH), 7.28 (d,J=4.8 Hz, 1H, ArH), 7.34(t,J=3.6 Hz, 1H, ArH), 7.76(d,J=4.4 Hz, 1H, g-H), 8.27(s, 1H, c-H); ESI-MSm/z: 249(M++1, 100%)。
5j: 褐色晶体,m.p.145.7 ℃~146.9 ℃;1H NMR(CDCl3)δ: 0.88(s, 6H, CH3), 1.43(q,J=8.0 Hz, 2H, f-H), 1.55~1.60(m, 1H, d-H), 2.46(t,J=8.0 Hz, 2H, e-H), 6.84(d,J=4.8 Hz, 1H, h-H), 7.41(d,J=4.8 Hz, 1H, g-H), 7.86(s, 1H, c-H); ESI-MSm/z: 223(M++1, 100%)。
[1] Schlapbach A, Feifel R, Hawtin S,etal. Pyrrolo-pyrimidones:A novel class of MK2inhibitors with potent cellular activity[J].Bioorg Med Chem Lett,2008,18:6142-6146.
[2] Muraglia E, Kinzel O D, Gardelli C,etal. Design and synthesis of bicyclic pyrimidinones as potent and orally bioavailable HIV-1 integrase inhibitors[J].J Med Chem,2008,51:861-874.
[3] Chereze N, Gallet T, Lochead A,etal. Preparation of arylamide pyrimidone derivatives for the treatment of neurodegenerative diseases[P]. WO 2 008 155 669,2008.
[4] Aquila B, Block M H, Davies A,etal. Preparation of novel pyrimidone-containing fused heterocycles as Eg5 inhibitors for treating neoplasm[P]. WO 2 004 078 758,2004.
[5] Bartroli O J, Anguita L M. Method for preparing pyrimidone derivatives with antifungal activity, e.g.,UR-9825[P].WO 2 001 066 519,2001.
[6] Cebasek P, Wagger J, Bevk D,etal. Parallel solution-phase synthesis of (Z)-3-(arylamino)-2,3-dehydroalanine derivatives and solid-phase synthesis of fused pyrimidones[J].J Comb Chem,2004,6:356-362.
[7] Kocar T, Recnik S, Svete J,etal. Transformations of 3-aminopyridazines.Synthesis of 4-oxo-4H-pyrimido[1,2-b]pyridazine and 1-(substituted pyridazin-3-yl)-1H-1,2,3-triazole derivatives[J].ARKIVOC,2002,8:143-156.
[8] Toplak R, Svete J, Grdadolnik S G,etal. Methyl (Z)-2-[(benzyloxycarbonyl)amino]-3-dimethyl-aminopropenoate in the synthesis of heterocyclic systems.Synthesis of (benzyloxycarbonyl)amino substituted fused pyrimidinones[J].Collect Czech Chem Commun,1999,64(2):177-189.
[9] Ma G, Jiang J, Shi M,etal. Recent extensions of the Morita-baylis-hillman reaction[J].Chem Commun,2009:5496-5514.
[10] Basavaiah D, Rao P D, Hyma R S. The Baylis-hillman reaction:A novel carbon-carbon bond forming reaction[J].Tetrahedron,1996,52(24):8001-8062.
[11] Singh V, Batra S. Advances in the Baylise-hillman reaction-assisted synthesis of cyclic frameworks[J].Tetrahedron,2008,64:4511-4574.
[12] Basavaiah D, Reddy B S, Badsara S S. Recent contributions from the Baylis-hillman reaction to organic chemistry[J].Chem Rev,2010,110:5447-5674.
[13] Zhong W, Liu Y, Wang G,etal. Recent advances in construction of nitrogen-containing heterocycles from Baylis-hillman adducts[J].Org Prep Proced Int,2011,43:1-66.
[14] Zhong W, Guo B, Lin F,etal. Regioselective synthesis of 5H-thiazolo[3,2-a]pyrimidin-5-ones from Morita-baylis-hillman adduct acetates under solvent-free and base-free conditions[J].Synthesis,2009,(10):1615-1622.
猜你喜欢
中学生数理化·高一版(2022年4期)2022-05-09
云南化工(2021年6期)2021-12-21
昆明医科大学学报(2021年8期)2021-08-13
科学(2020年2期)2020-08-24
武警医学(2018年10期)2018-11-06
浙江大学学报(工学版)(2016年9期)2016-06-05
当代化工研究(2016年6期)2016-03-20
当代化工研究(2016年5期)2016-03-20
生物技术通报(2015年1期)2015-04-10
无机化学学报(2014年3期)2014-02-28
