
两种天然异戊烯基黄烷酮的全合成*
2013-03-26 06:02郭冬冬杨金会梁西周落俊山黄文倩
合成化学 2013年5期
郭冬冬,杨金会,梁西周,落俊山,黄文倩
(1.宁夏大学天然气转化国家重点实验室培育基地,宁夏银川 750021;2.南京先声东元制药有限公司,江苏南京 211800)
黄烷酮在植物中广泛存在,其中大部分均具有广泛的生理和药理活性[1~4]。其中异戊烯基黄烷酮在黄酮类化合物的研究中占有重要地位,由于异戊烯基的存在,常常使其具有显著的生理及药理活性[5]。
(±)-5,3'-二羟基-7,8-(2,2-二甲基吡喃)-4'-甲氧基黄烷酮(1)是英国科学家 B.Md.Mukhlesur Rahman等[6]于 2001 年从植物象橘(Feronia limonia)的茎杆中提取分离得到的一种新的苯并吡喃型黄烷酮,有很好的抗菌活性。(± )-5,7,3'-三羟基-4'-甲氧基-8-异戊烯基-黄烷酮(2)是美国科学家 Muhammad Ali Versiani等[7]于2011年从Macaranga conifera中提取分离得到的一种新的异戊烯基黄烷酮天然产物,其抑制ABCG2 的 IC50值为6.6 μM。
本课题组一直关注异戊烯基黄酮类化合物的研究,并合成了一些天然产物[8~11]。作为这一工作的继续,本文以3-甲氧甲氧基-4-甲氧基苯甲醛(5)和 2,4,6-三羟基-3-异戊烯基苯乙酮(3)为原料,经烷基化和缩合反应合成了新化合物2'-羟基-4-甲氧基-3,4',6'-三甲氧甲氧基-3'-异戊烯基査尓酮(6);6经环合反应合成了新化合物(±)-5,7,3'-三甲氧甲氧基-4'-甲氧基-8-异戊烯基黄烷酮(7);7经脱保护和环合反应实现了1和2的全合成(Scheme 1),总收率分别为11.6%和10.5%,其中1为首次合成,其结构经1H NMR,IR和MS表征。
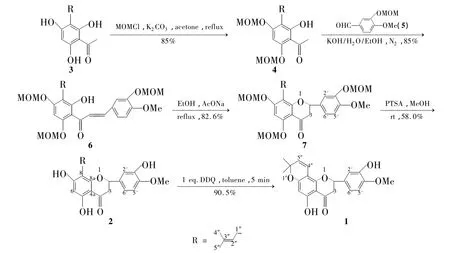
Scheme 1
1 实验部分
1.1 仪器与试剂
X-5型熔点仪(温度未校正);Bruker AM-400 MHz型核磁共振仪(CDCl3为溶剂,TMS为内标);FT-IR 8430S型红外光谱仪(KBr压片);Thermo LTQ-XL型质谱仪。
3[13]和 5[12,16]按文献方法合成[3:淡黄色固体,收率34.9%,m.p.166 ℃ ~167 ℃;1H NMR(DMSO)δ:1.69(s,3H,CH3),1.61(s,3H,CH3),2.56(s,3H,COCH3),3.09(d,J=6.8 Hz,2H,CH2),5.14(t,J=6.8 Hz,1H,CH=),6.01(s,1H,ArH),10.51(s,1H,OH),10.28(s,1H,OH),14.03(s,1H,OH);IR ν:3 417,3 321,1 629,1 434,1 284,1 072,817 cm-1。5:无色油状液体,收率 96%;1H NMR δ:3.88(s,3H,OCH3),3.44(s,3H,OCH3),5.20(s,2H,OCH2O),6.93(d,J=8.4 Hz,1H,ArH),7.46(d,J=8.4 Hz,1H,ArH),7.58(s,1H,ArH),9.76(s,1H,COH);IR ν:2 956,1 697,1 267,1 000,811 cm-1];300目 ~400目和 GF254硅胶,青岛海洋化工厂;其余所用试剂均为分析纯,其中丙酮用前经除水处理。
1.2 合成
(1)2-羟基-4,6-二甲氧甲氧基-3-异戊烯基苯乙酮(4)的合成[13]
在反应瓶中加入3 578 mg(2.5 mmol)和丙酮10 mL,搅拌使其溶解;剧烈搅拌下加入无水碳酸钾910 mg(6.6 mmol),回流反应 10 min;用注射器缓慢滴加氯甲基甲基醚(MOMCl)0.58 mL(6.0 mmol),滴毕,回流反应2 h。减压蒸除溶剂,残余物加水溶解,用乙酸乙酯(3×30 mL)萃取,合并有机相,依次用蒸馏、饱和食盐水洗涤,无水硫酸镁干燥,减压蒸除溶剂后经硅胶柱层析[洗脱剂:A=V(石油醚)∶V(乙酸乙酯)=4∶1]纯化得淡绿色液体 5 675 mg,收率 85%;1H NMR δ:1.77(s,3H,CH3),1.65(s,3H,CH3),2.65(s,3H,COCH3),3.30(d,J=6.8 Hz,2H,CH2),3.47(s,3H,OCH3),3.50(s,3H,OCH3),5.19(m,1H,CH=),5.23(s,2H,OCH2O),5.24(s,2H,OCH2O),6.38(s,1H,ArH),13.82(s,1H,OH);IR ν:3 425,2 952,1 623,1 419,1 272,1 120,892 cm-1。
(2)6 的合成[14]
在反应瓶中加入5 423 mg(2.2 mmol),4 610 mg(1.8 mmol)和乙醇9 mL,搅拌下于0℃缓慢滴0℃混合液[氢氧化钾5.6 g(100 mmol)+水5.4 mL+乙醇8 mL],氮气保护下于0℃反应1 h;于室温反应24 h。倒入冰水中,用3 mol·L-1盐酸调至pH<2,用二氯甲烷(3×7 mL)萃取,合并有机相,依次用蒸馏水、饱和食盐水洗涤,无水硫酸镁干燥,蒸除溶剂后经硅胶柱层析(洗脱剂:A=5∶1)纯化得桔红色固体6 630 mg,收率85%,m.p.112 ℃ ~ 114 ℃;1H NMR δ:1.67(s,3H,CH3),1.79(s,3H,CH3),3.33(d,J=7.2 Hz,2H,CH2),3.48(s,3H,OCH3),3.53(s,3H,OCH3),3.58(s,3H,OCH3),3.92(s,3H,OCH3),5.22(m,1H,CH=),5.24(s,2H,OCH2O),5.26(s,2H,OCH2O),5.28(s,2H,OCH2O),6.41(s,1H,ArH),6.91(d,J=8.4 Hz,1H,ArH),7.22(dd,J=2.0 Hz,8.4 Hz,1H,ArH),7.51(d,J=2.0 Hz,1H,ArH),7.74(d,J=15.6 Hz,1H,βCH=),7.83(d,J=15.6 Hz,1H,αCH=),13.91(s,1H,OH);IR ν:3 566,3 162,2 952,2 360,1 357,1 182,1 510,1 264,1 075,971 cm-1;MSm/z:501{[M -1]+}。
(3)7 的合成[14]
在反应瓶中加入6 502 mg(1.0 mmol)和乙醇16.0 mL,搅拌使其溶解;加入无水醋酸钠410 mg(5 mmol)和五滴水,回流反应25 h。冷却至室温,加少量水,用二氯甲烷(3×10 mL)萃取,合并有机相,依次用蒸馏、饱和食盐水洗涤,无水硫酸镁干燥,蒸除溶剂后经硅胶柱层析(洗脱剂:A=3∶1)纯化得淡黄色黏稠液体 7 414 mg,收率82.6%;1H NMR δ:1.64(s,6H,CH3),2.79(dd,J=3.2 Hz,16.4 Hz,1H,3-Hβ),2.99(dd,J=12.8 Hz,16.8 Hz,1H,3-Hα),3.30(d,J=6.4 Hz,2H,CH2),3.47(s,3H,OCH3),3.52(s,3H,OCH3),3.90(s,3H,OCH3),3.92(s,3H,4'-OCH3),5.19(m,1H,=CH),5.23(s,2H,CH2O),5.24(s,2H,CH2O),5.25(s,2H,CH2O),5.33(dd,J=2.8 Hz,12.8 Hz,1H,2-H),6.56(s,1H,ArH),6.92(d,J=8.4 Hz,1H,ArH),7.07(dd,J=2.0 Hz,8.4Hz,1H,ArH),7.28(d,J=2.0 Hz,1H,ArH);IR ν:3 775,3 545,3 105,2 926,2 468,2 360,1 516,1 600,1 547,1 413,1 154,695 cm-1;MSm/z:501{[M -1]+}。
(4)2的合成
在反应瓶中加入7 104 mg(0.2 mmol)和甲醇4 mL,搅拌均匀;加对甲苯磺酸(PTSA)114 mg(0.6 mmol),于室温反应36 h(TLC检测)。加水5 mL,用乙酸乙酯(3×15 mL)萃取,合并有机相,依次水、饱和食盐洗涤,无水硫酸钠干燥,减压蒸除溶剂后经硅胶柱层析[洗脱剂:B=V(石油醚)∶V(丙酮)=3∶1]纯化得白色粉末2 43 mg,收率58.0%,m.p.158 ℃ ~162 ℃;1H NMR(氘代甲醇)δ:1.58(s,3H,CH3),1.61(s,3H,CH3),2.70(dd,J=3.2 Hz,16.8 Hz,1H,3-Hβ),3.02(dd,J=12.8 Hz,16.8 Hz,1H,3-Hα),3.15(dd,J=7.0 Hz,14.0,1H,1″-H),3.20(d,J=7.6 Hz,14.4 Hz,1H,1″-H),3.86(s,3H,OCH3),5.13(m,1H, =CH),5.28(dd,J=3.2 Hz,12.4 Hz,1H,2-H),5.92(s,1H,ArH),6.90(dd,J=2.0 Hz,8.4 Hz,1H,ArH),6.93(d,J=8.4 Hz,1H,ArH),6.96(d,J=2.0 Hz,1H,ArH);13C NMR(氘代甲醇)δ:198.0(C4),166.1(C5),163.1(C7),161.5(C8a),149.3(C4'),147.8(C3'),133.5(C1'),131.7(C3″),123.9(C2″),118.9(C6'),114.6(C2'),112.5(C5'),109.1(C8),103.4(C4a),96.4(C6),80.1(C2),56.5(C4'-OMe),44.0(C3),26.0(C5″),22.5(C1″),17.9(C4″);IR ν:3 009,3 474,3 092,2 917,2 642,2 509,1 864,1 623,1 548,1 269,1 205,1 079,1 042,905,808,764,736 cm-1;MSm/z:369{[M -1]+}。
(5)1的合成
在反应瓶中加入2 37 mg(0.1 mmol)和甲苯2 mL,搅拌使其溶解;加入二氯二腈基苯醌(DDQ)23 mg(0.1 mmol),回流反应 5 min(TLC跟踪)。加少量水,用乙酸乙酯(3×3 mL)萃取,合并有机相,依次用蒸馏、饱和食盐水洗涤,无水硫酸钠干燥,减压蒸除溶剂后经硅胶柱层析(洗脱剂:B=4∶1)纯化得黄色固体1 33 mg,收率90.5%;1H NMR δ:1.42(s,3H,CH3),1.46(s,3H,CH3),2.80(dd,J=17.0 Hz,3.0 Hz,1H,3-H),3.05(dd,J=13.2 Hz,17.2 Hz,1H,3-H),3.93(s,3H,4'-OCH3),5.33(dd,J=2.4 Hz,12.8 Hz,1H,2-H),5.46(d,J=10 Hz,1H,5″-H),5.99(d,J=0.4 Hz,1H,6-H),6.54(dd,J=10 Hz,0.4 Hz,1H,4″-H),6.89(d,J=8.0 Hz,1H,5'-H),6.93(dd,J=2.0 Hz,8.4 Hz,1H,5'-H),7.06(d,J=2.0 Hz,1H,2'-H),12.07(s,1H,5-OH);13C NMR δ:196.0(C4),163.9(C5),162.4(C7),156.9(C9),147.0(C4'),146.0(C3'),131.8(C1'),126.6(C3″),118.3(C2'),115.8(C4″),112.8(C5'),110.7(C6'),103.0(C10),102.1(C8),97.7(C6),78.9(C2),78.3(C6″),56.2(C4'-OMe),43.3(C3),28.7,28.4(C6″-Me);IR ν:2 917,1 708,1 628,1 510,1 362,1 215,523 cm-1;EI-MSm/z(%):368(21),353(56),337(4),217(4),203(100),135(22),77(22);HR-MS:Calcd for C21H20O6[M+]368.126 0,found 368.125 8。波谱数据与文献[6]值一致。
2 结果与讨论
2.1 合成
脱除MOM 保护基,常常使用3 mol·L-1盐酸在甲醇中回流脱除。本文在7的合成中,采用3 mol·L-1盐酸在甲醇中回流脱除,效果不好,估计由于HCl酸性太强,脱除MOM保护基后羟基随即和邻位的乙戊烯基发生环合。实验中改用PTSA在甲醇中反应,以58.0%的收率得2。
[1]Daskiewicz J B,Depeint F,Viornery L,et al.Effects of flavonoids on cell proliferation and caspase activation in a human colonic cell line HT29:An SAR study[J].J Med Chem,2005,48:2790 -2804.
[2]Comte G,Daskiewicz J B,Bayet C,et al.Rearrangement of 5-O-prenyl flavones:A regioselective access to 6-C-(1,1-dimethylallyl)-and 8-C-(3,3-dimethylallyl)-flavones[J].J Med Chem,2001,44:763 -768.
[3]Na M,Jang J,Njamen D,et al.Prenylated flavonoids with PTP1B inhibitory activity from the root bark of Erythrinam ildbraedⅡ[J].J Nat Prod,2006,69:1572 -1576.
[4]Harborne J B,Williams C A.Anthocyanins and other flavonoids[J].J Nat Prod Rep,2001,18:310 -333.
[5]Nikaido T,Ohmoto T,Kinoshita T,et al.Inhibition of adenosine 3',5'-cyclic monophosphate phosphodiesterase by flavonoids Ⅲ[J].Chem Pharm Bull,1989,37:1392-1395.
[6]Rahman M M,Gray A I.Antimicrobial constituents from the stem bark of Feronia limonia[J].Phytochemistry,2002,59:73 -77.
[7]Muhammad Ali Versiani,Thushara Diyabalanage,Ranjala Ratnayake,et al.Flavonoids from eight tropical plant species that inhibit the multidrug resistance transporter ABCG2[J].J Nat Prod,2011,74:262 -266.
[8]Li Y,Yang J H,Li W D Z.Facile synthesis of(±)-7-hydroxy-3',4'-methoxylenedioxy flavan and(± )-4'-hydroy-7-methoxy flavan by a BF3-Et2O-mediated pyrancyclization[J].J Nat Prod,2001,64:214 -216.
[9]Yang J H,Zhao Y M,Ji C B.First total synthesis of(± )-abyssinoflavanone V[J].Chin Chem Lett,2008,19:658-660.
[10]杨金会,孟丽聪.天然产物1,3-二(2-羟基-4-甲氧基苯基)丙烷和1,3-二-(2,4-二羟基苯基)丙烷的合成[J].有机化学,2008,28:918 -921.
[11]Zhang Y H,Yang J H,Li H J,et al.First total synthesis of(±)-puyanin and(±)-4'-O-methylbonannione[J].Chin J Chem,2011,29:521 -524.
[12]薛吉军.黄酮类化合物的合成[D].兰州:兰州大学,2004.
[13]孙亚捷,李裕林,赵联运,等.4',5,7-三羟基-2'-甲氧基-6,8-二异戊烯基黄烷酮的全合成[J].合成化学,1995,(02):127 -130.
[14]C S Huang,Z Zhang,Y L Li,et al.Total synthesis of(R,S)-sophoraflavanone C[J].J Nature Product,1998,61:1283 -1285.
[15]落俊山,杨金会,王金荣,等.Paratocarpin B的全合成[J].合成化学,2012,20(5):578 -581.
[16]黄文倩,杨金会,郭冬冬,等.3″,3″-二甲基吡喃[3',4']2,4,2'-三羟基查尔酮的首次全合成[J].合成化学,21(1):050-052.
猜你喜欢
中国药业(2020年7期)2020-04-11
吉林农业(2019年6期)2019-06-11
天然产物研究与开发(2018年10期)2018-11-06
天然产物研究与开发(2018年10期)2018-11-06
中成药(2017年10期)2017-11-16
中成药(2017年4期)2017-05-17
中国农业科学(2017年5期)2017-03-22
中国医学科学院学报(2015年5期)2015-03-01
合成化学(2014年2期)2014-06-23
天然产物研究与开发(2014年6期)2014-04-27
