
苯磺酰胺从碳酸酐酶ll中脱离过程的分子动力学模拟
2013-07-25 09:12孙维琦张继龙郑清川孙志伟张红星
物理化学学报 2013年4期
孙维琦 张继龙 郑清川,* 孙志伟 张红星
(1吉林大学公共卫生学院,长春 130021;2吉林大学理论化学研究所,理论化学计算国家重点实验室,长春 130023;3北华大学公共卫生学院,吉林吉林 132013;4首都医科大学公共卫生与家庭医学院,北京 100069)
1 引言
二氧化碳是人体细胞内糖类和脂肪代谢的副产物,必须及时从体内清除.碳酸酐酶是与这一过程密切相关的一类重要含锌金属酶,它能够催化二氧化碳和碳酸氢根离子的相互转化.1虽然该反应在无酶条件下也能发生,但碳酸酐酶将这个反应的转化效率提高了一百万倍.2人体内大约70%的二氧化碳都是在这类酶的作用下代谢排出体外的.3除此之外,碳酸酐酶参与多种重要的生理过程,包括酸碱平衡,4骨骼的生长和功能,5代谢,6肿瘤胞外环境的酸化,7信号传导和记忆等.8-10
碳酸酐酶II(CAII)是研究最广泛的一种碳酸酐酶异构酶,它大量存在于红血球中,具有广泛的组织分布.这个异构酶也是目前已知的所有哺乳动物碳酸酐酶中最具有催化效率的,拥有接近扩散速度的催化速率.11除了代谢二氧化碳外,CA II催化的碳酸氢根的生成对于维持眼内晶状体压力的平衡也具有重要的生理作用.12,13同时,骨骼石化症14和绝经期后的骨质疏松症也都和CAII的功能紊乱相关.
芳基磺胺类药物一直被作为重要的碳酸酐酶抑制剂,许多临床上应用的药物都是通过苯磺酰胺作为母体发挥作用的(图1).15尽管对这类药物的研究已十分充分,但却很少有研究关注它与碳酸酐酶的具体结合过程.在这方面,分子动力学模拟方法具有独特的优势.16-19因此,本文以最基本的苯磺酰胺分子作为底物,通过多种分子动力学方法的综合运用,从理论上探索苯磺酰胺分子与CA II的具体结合过程及原子水平上的相互作用,确定影响动态结合的重要氨基酸残基.由于CA II具有整个碳酸酐酶家族的许多共性特征,因此当前的底物结合研究将具有普遍的意义.
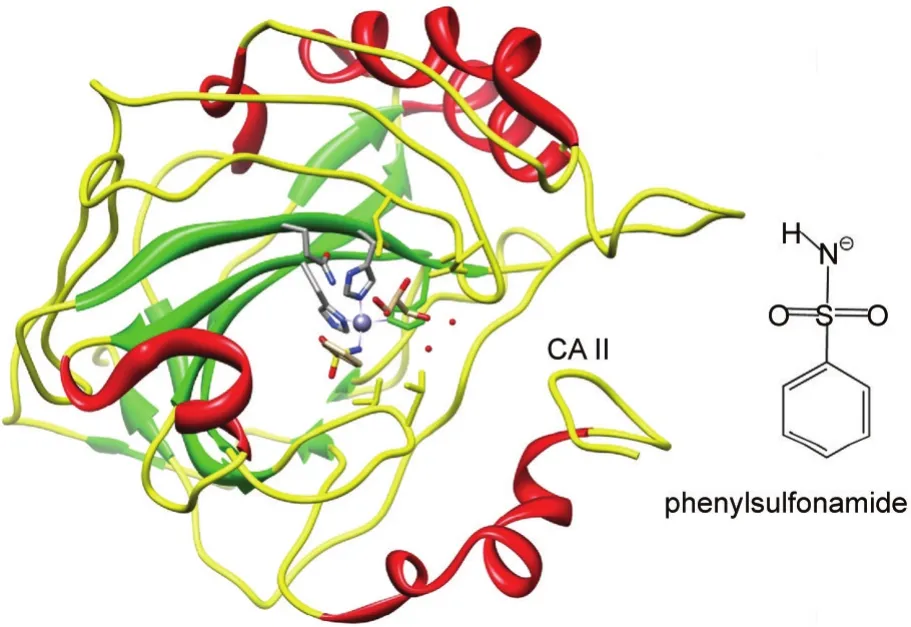
图1 碳酸酐酶II及其抑制剂苯磺酰胺的结构Fig.1 Molecular structures of carbonic anhydrase II and its inhibitor(phenylsulfonamide)
2 计算方法
2.1 常规分子动力学模拟
实验上已经获得了碳酸酐酶II复合小分子磺胺抑制剂的X射线衍射晶体结构,蛋白质结构数据库(www.rcsb.org)收录号为2WEJ.该结构中包含了碳酸酐酶II(含有258个氨基酸残基),一个锌离子,基本磺胺单元,一个甘油分子和若干结晶水.甘油分子在模拟中被删除,其它部分都保留.为了获得稳定的复合物结构并进行充分的构象空间取样,首先执行常规的分子动力学模拟.分子结构中丢失的氢原子利用VMD程序20补充,然后利用其中的Solvate插件为复合物添加一个长方体的TIP3P溶剂水盒子,保证溶质与水盒子边界的最小距离不低于1 nm.浓度为154 mmol·L-1的Na+和Cl-被加入体系中以模拟生理条件同时中和体系自身的电荷.
采用PME方法21计算长程静电相互作用,范德华相互作用在1.2-1.4 nm范围内逐渐减小为0.采用周期性边界条件以模拟连续一致的行为.首先对整个体系进行10000步的共轭梯度(CG)能量最小化,该过程采用4184 kJ·mol-1·nm-2的谐振子势函数限制蛋白骨架和磺胺分子的位置.然后保持限制,采用Langevin控温法22将系统的温度以5 ps·K-1的速度从100 K逐渐加热到300 K,模拟时间步长设为1 fs.此后在1 ns的时间内将限制势逐渐减小到0.最后控制压力为1.01325×105Pa,在NPT系综下对体系进行10 ns的平衡模拟.最终得到的稳定构象被用于下一步的拉伸分子动力学计算.
2.2 拉伸分子动力学模拟及平均力势的计算
拉伸分子动力学(SMD)方法是一种特殊的分子动力学模拟方法,能够模拟配体-受体的脱离或结合过程,已成功应用于许多相关研究中.20,21该方法可分为两类:常速SMD和常力SMD.在本文中使用的是前者.常速SMD方法的基本原理是在配体的某一点上附着一个虚拟弹簧,弹簧另一端与一个无质量的虚拟原子相连.在模拟过程中,使虚拟原子以设定的恒速移动,虚拟弹簧产生形变,其弹力即可反映配体-受体间的相互作用.在上述过程中,弹力F(t)可通过下式求得:

其中,k代表劲度系数,v是虚拟原子的移动速度,x是作用点相对于初始位置的位移.
对于当前的研究体系来说,基于常规分子动力学模拟获得稳定复合构象,我们固定金属锌离子和三个蛋白配位残基(His94、His96和His119),将虚拟弹簧附着在磺胺小分子的质心上,沿着底物与锌的配位键方向进行拉伸模拟.动力学模拟步长为1 fs,轨迹每0.5 ps保存一次,拉力值每20 fs记录一次.使用不同的随机种子,拉伸模拟被重复三次进行验证.
为了研究苯磺酰胺脱离过程的能量变化,我们基于SMD模拟获得的脱离路径上的特定结构计算了沿脱离路径的自由能变化.反应坐标选为苯磺酰胺质心与配位Zn2+之间的距离(0.4-1.8 nm).NAMD(nanoscalemoleculardynamics)中的自洽偏置力(ABF)方法23被用于计算对应于该反应坐标的平均力势(PMF).为了提高计算效率,整个反应坐标被分成长度为0.1 nm的若干个窗口.在每个窗口内都运行长达2 ns的取样模拟.最终得到的各段能量数据再通过一个附加的模拟进行联合.
3 结果与讨论
3.1 复合体系的稳定性
当前研究所用的初始模型来源于X射线衍射晶体结构,其中必然存在着局部高势能构象,而这些构象若不进行充分平衡将会影响接下来的SMD模拟.因此,碳酸酐酶II-磺胺复合体系首先经过了10 ns的常规分子动力学模拟.蛋白的结构稳定性是通过计算均方根偏差(RMSD)来表征的.图2所示的就是10 ns的NPT模拟过程中,碳酸酐酶II的全原子以及骨架原子的RMSD随模拟时间变化的曲线.从图中可以看出,在该段模拟开始后的很短时间内,二者就分别稳定在0.16、0.08 nm附近并做小幅度波动,证明蛋白的整体结构已经稳定.此外,为了证明局部结构的稳定性,当前研究也考察了金属配位键的键长随模拟时间的变化.结果表明,包括磺胺分子(通过磺酰基的氮原子与锌配位)在内的四个配位键键长在NPT过程中保持很好的稳定性且与晶体结构中的键长十分接近,证明局部结构也是稳定的.
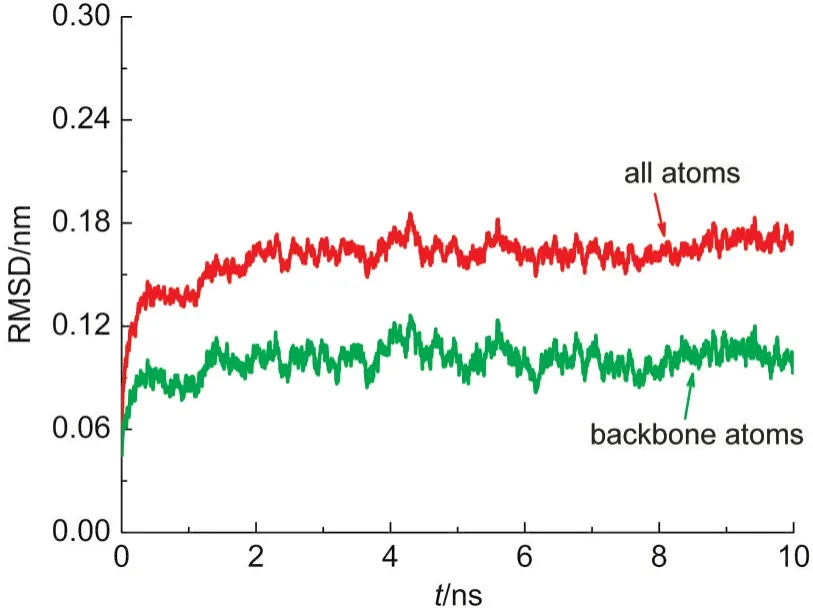
图2 10 ns NPT模拟过程中碳酸酐酶II的全原子和骨架原子的均方根偏差(RMSD)Fig.2 Root mean square deviation(RMSD)profiles of all atoms and backbone atoms of CAII in the 10 ns NPT simulation
3.2 拉伸分子动力学模拟及PMF的计算
基于上述获得的稳定复合物构象,拉伸分子动力学模拟被用于研究磺胺小分子从碳酸酐酶II中脱离的过程.对于拉伸分子动力学模拟来说,拉伸方向、拉伸速度和虚拟弹簧的劲度系数都对最终的拉力曲线有直接的影响.24-26磺胺小分子与碳酸酐酶II的结合构象显示,底物的脱离不存在多种不同的路径,我们的模拟测试也证实了方向的差异对拉力曲线的影响不大.关于拉伸速度和劲度系数,结合已有的类似模拟,16,24-26当前研究分别考察了三种不同的劲度系数(5000、350和1 pN·nm-1)和两种不同拉伸速度(0.2和0.5 nm·ns-1)下得到的拉力曲线.不同的劲度系数会影响拉力曲线的表现并给出不同大小的峰值.当劲度系数为5000 pN·nm-1时,拉力曲线表现出很大的波动,导致拉力峰值难以分辨,并且拉力曲线的重现性不好;而1 pN·nm-1的劲度系数导致到达拉力峰值的时间变长,从而间接延长了模拟时间,因而也不适合当前模拟.处于二者之间的劲度系数(350 pN·nm-1)既可以保证较快的拉力增速,降低模拟时长,又能够适度地消除拉力曲线的波动,给出很好的拉力重现性.在这个劲度系数下,两个拉伸速度也被分别进行测试.当拉伸速度较小时(0.2 nm·ns-1),拉力峰值出现的时间较晚,模拟时间相对较长(超过10 ns);而0.5 nm·ns-1的拉伸速度可以在合理的时间内模拟出整个脱离过程,因而是较合适的拉伸速度.
在350 pN·nm-1的劲度系数和0.5 nm·ns-1的拉伸速度下,当前研究对磺胺小分子底物从碳酸酐酶II中脱离的过程进行了三次重复的SMD模拟,每次选择不同的随机数以确保统计结果的科学性,所得的拉力曲线如图3所示.从图中可以看出,三条曲线彼此重合得很好,大约在8.8 ns附近,三条曲线达到了各自的峰值,分别为1438、1419和1421 pN,三者相对平均值表现出了小于1%的波动幅度,显示了几乎相同的脱离力.峰值之后,拉力曲线迅速降低,此时磺胺小分子脱离了碳酸酐酶II的活性位点进入水溶液环境中,拉力曲线在0附近上下波动,表现出底物分子在水中的受力情况.
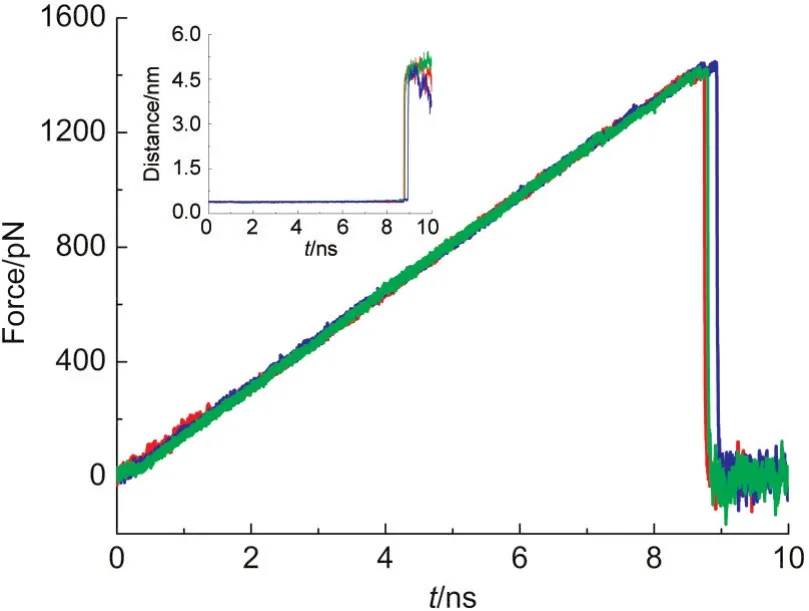
图3 拉伸分子动力学模拟过程中拉力随时间的变化图谱Fig.3 Time dependence of the applied force in the steered molecular dynamics(SMD)simulation
脱离过程中的自由能变化能够反映出配体与受体相互作用的动态变化情况,给出脱离时的许多细节信息.基于SMD模拟的轨迹,我们计算得到了沿着上述反应坐标的PMF变化图(见图4).图中显示,从反应路径的初始点,能量曲线即迅速上升,如此急剧的自由能消耗主要用于克服底物与金属锌的配位作用.直到反应坐标达到0.50 nm时,能量的消耗达到了95 kJ·mol-1.此后能量曲线开始下降,在0.59 nm时达到一个局部极小值点,随后又逐步升高,最后基本稳定下来.从PMF模拟轨迹上观察,底物开始进入溶剂是在反应坐标0.59 nm处,此时底物基本脱离活性位点,但尚未完全进入水环境中,而仅仅是一部分与水发生相互作用.在PMF曲线中,0.59 nm后的曲线基本反映了底物小分子的溶解过程.故此,这个极小值点是底物脱离过程中一个特殊的结合状态.对该点底物与蛋白相互作用的研究能够揭示影响底物结合的重要残基的功能.

图4 沿着底物脱离反应坐标的PMF变化图谱Fig.4 PMF profile along the reaction coordinate during the substrate's unbinding
3.3 磺胺小分子与酶的重要相互作用
磺胺小分子脱离碳酸酐酶II的整个过程已经通过SMD模拟及PMF的计算清晰地呈现出来,这为研究脱离路径上的关键氨基酸残基提供了可能.通过考察轨迹,当前研究详细分析了整个脱离过程中底物小分子和碳酸酐酶II重要残基的相互作用情况.首先,整个底物脱离过程中小分子与酶总相互作用能的计算结果显示,静电相互作用能对总相互作用能的贡献占80%左右.这表明,静电相互作用占据主导地位,能够极大地影响小分子与酶的结合.这与小分子的性质和酶的活性位点密切相关.当前研究中选取的底物是苯磺酰胺分子,这个分子的氨基上带有一个负电荷(图1),与碳酸酐酶II活性位点处的金属锌离子以及底物附近的带电残基之间存在着强烈的静电力,从而导致了静电作用远远大于范德华相互作用.

图5 苯磺酰胺分子和三个重要残基之间的总相互作用能Fig.5 Profiles of the total interaction energy between phenylsulfonamide molecule and three important residues
联合上述的PMF计算结果,所有影响底物与酶结合的重要氨基酸残基也通过相互作用能的计算来进行判断.在底物脱离路径上的所有氨基酸残基中,Leu198、Thr199和Thr200是三个最重要的残基.相互作用能的计算显示,这三个残基与苯磺酰胺之间的相互作用能远远大于其它路径上的残基,并且都是以静电作用为主导的.图5给出的是它们与小分子相互作用能随着SMD模拟时间的变化图谱.从中可以看出,在三个残基中,Leu198与底物的相互作用在SMD模拟的大部分时间内远小于另外两个残基且多有不利于底物结合的情况发生.但这个残基在底物分子即将脱离碳酸酐酶II的时刻,与其相互作用急剧降低,阻碍了底物的离去,因此是影响底物结合的一个重要残基.而对于Thr199和Thr200来说,二者在大多数情况下均和苯磺酰胺分子存在很强的相互作用,其中以Thr200作用更强.虽然在底物分子脱离时刻这两个残基没有出现如Leu198那样的强烈阻碍作用,但因为它们与底物长时间较大的相互作用,所以也是影响底物结合或脱离的关键氨基酸残基.除了上述这三个残基外,其它一些残基与底物分子也有较强的作用.例如,Asn62和Ala65,这两个残基在底物脱离时刻与之的相互作用也分别达到了-27.2和-19.2 kJ·mol-1,并且二者也都是以静电作用为主的.此外,活性位点锌离子的配位残基(His94,His96和His119)则由于静电作用对底物的脱离起到促进作用.与底物存在较强范德华相互作用的残基是Val121,对于这个残基来说,静电作用和范德华相互作用在数值上相差不大,都在-8.4 kJ·mol-1附近,由此可见,这个残基与底物的苯环之间存在较强的作用.
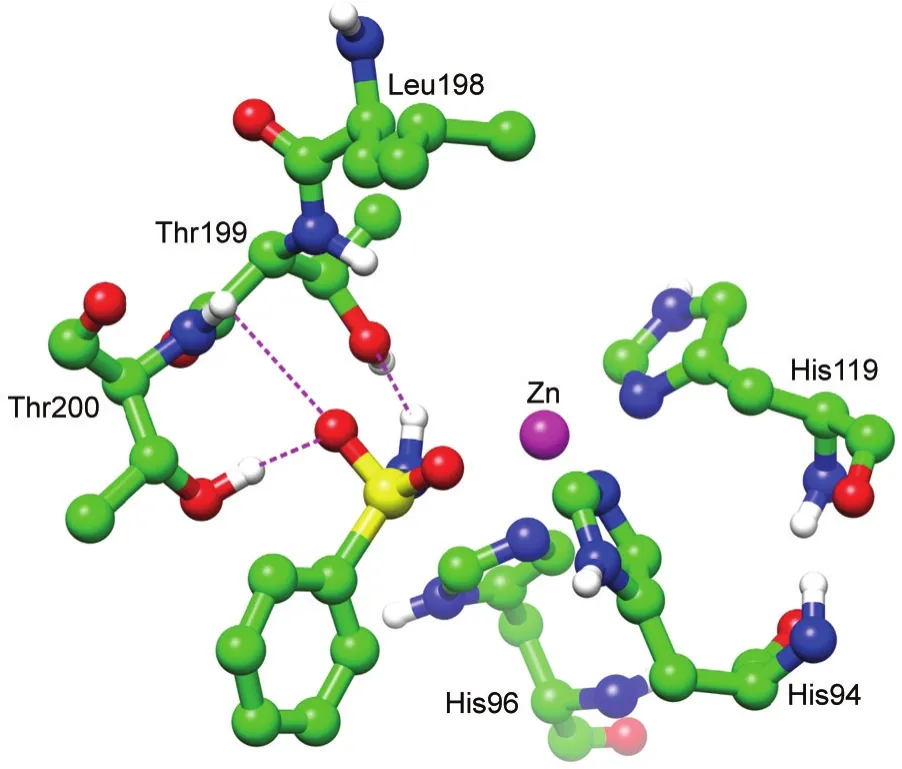
图6 底物脱离过程中苯磺酰胺分子和重要残基之间的原子水平相互作用情况Fig.6 Atomic level interaction between phenylsulfonamide and the important residues in the process of the substrate
能量的计算不能揭示底物分子脱离碳酸酐酶II时原子水平上的作用情况.因此,当前研究详细考察了PMF模拟轨迹中底物与酶的作用,其中脱离过程关键点的相互作用情况如图6所示.在这一位置处(对应于PMF曲线的波谷点),苯磺酰胺分子与酶残基存在多条氢键,分别为底物氨基氢与Thr199的支链羟基氧之间的一个氢键和底物磺酰基氧与残基Thr200的支链羟基氢和主链氨基氢之间的两个氢键,而这个磺酰氧虽然没有与Thr199的主链氨基氢形成氢键,但两个原子之间的距离并不太远,因此可以断定它们之间的相互作用也是相当强的.而苯磺酰胺分子与Leu198之间的相互作用则发生在模拟的后续阶段,主要是磺酰氧与这个残基主链氨基氢之间的作用.
对模拟轨迹的考察也揭示了磺胺小分子底物脱离碳酸酐酶II整个过程中的酶和小分子的变化.对于碳酸酐酶II来说,底物小分子脱离之前,其活性位点处的锌离子分别与His94、His96、His119和苯磺酰胺配位;底物脱离后,一个外来的水分子进入活性位点中,占据了原来配体的位置与锌离子形成配位键.碳酸酐酶II的其它残基没有发生较大的构象变化,这源于该酶较强的活性位点刚性特征.8对于底物小分子来说,脱离初始阶段它始终保持着磺酰基朝向活性位点方向,而其苯环则指向活性位点外的溶液环境.脱离后,由于磺酰基团的亲水性,底物发生翻转,苯环朝向活性位点方向,而磺酰胺部分朝向水溶剂,以利于磺酰基与溶剂水分子的作用.
4 结论
磺胺类药物分子与碳酸酐酶II的结合或脱离除了受到活性位点残基的影响外,那些在底物结合路径上的残基也起到不可忽视的作用.对于磺胺类抑制剂来说,以苯磺酰胺为例的模拟揭示了这类底物与酶之间的静电相互作用对于小分子与酶的结合起到关键角色.在结合路径上的三个关键氨基酸残基Leu198、Thr199和Thr200通过与底物的磺酰胺部分形成氢键,从而阻碍底物的离去.
(1)Krishnamurthy,V.M.;Kaufman,G.K.;Urbach,A.R.;Gitlin,I.;Gudiksen,K.L.;Weibel,D.B.;Whitesides,G.M.Chem.Rev.2008,108,946.doi:10.1021/cr050262p
(2) Sanyal,G.;Maren,T.H.J.Biol.Chem.1981,256,608.
(3) Stabenau,E.K.;Heming,T.Comp.Biochem.Physiol.Part A:Mol.Integr.Physiol.2003,136,271.doi:10.1016/S1095-6433(03)00177-6
(4) Shah,G.N.;Ulmasov,B.;Waheed,A.;Becker,T.;Makani,S.;Svichar,N.;Chesler,M.;Sly,W.S.Proc.Natl.Acad.Sci.U.S.A.2005,102,16771.doi:10.1073/pnas.0508449102
(5) Gay,C.V.;Weber,J.A.Crit.Rev.Eukaryotic Gene Expression2000,10,213.
(6) Henry,R.P.Ann.Rev.Physiol.1996,58,523.doi:10.1146/annurev.ph.58.030196.002515
(7) Svastova,E.;Hulikova,A.;Rafajova,M.;Zatovicova,M.;Gibadulinova,A.;Casini,A.;Cecchi,A.;Scozzafava,A.;Supuran,C.T.;Pastorek,J.;Pastorekova,S.FEBS Lett.2004,577,439.doi:10.1016/j.febslet.2004.10.043
(8) Sun,M.K.;Alkon,D.L.Trends Pharm.Sci.2002,23,83.doi:10.1016/S0165-6147(02)01899-0
(9) Scoozzafava,A.;Mastrolorenzo,A;Supuran,C.T.Expert Opin.Ther.Pat.2006,16,1627.doi:10.1517/etp.2006.16.issue-12
(10) Pastorekova,S.;Parkkila,S.;Pastorek,J.;Supuran,C.T.J.Enzyme Inhib.Med.Chem.2004,19,199.doi:10.1080/14756360410001689540
(11) Khalifah,R.G.J.Biol.Chem.1971,246,2561.
(12)Wistrand,P.J.;Schenholm,M.;Lonnerholm,G.Invest.Ophthalmol.Vision Sci.1986,27,419.
(13)Alward,W.L.M.N.Engl.J.Med.1998,339,1298.doi:10.1056/NEJM199810293391808
(14) Borthwick,K.J.;Kandemir,N.;Topaloglu,R.;Kornak,U.;Bakkaloglu,A.;Yordam,N.;Ozen,S.;Mocan,H.;Shah,G.N.;Sly,W.S.;Karet,F.E.J.Med.Genet.2003,40,115.doi:10.1136/jmg.40.2.115
(16)Zhang,J.L.;Zheng,Q.C.;Li,Z.Q.;Zhang,H.X.PLoS ONE2012,7,e39546.
(17) Dong,X.Y.;Du,W.J.;Liu,F.F.Acta Phys.-Chim Sin.2012,28,2735.[董晓燕,都文婕,刘夫锋.物理化学学报,2012,28,2735.]doi:10.3866/PKU.WHXB201207162
(18)Zhao,Y.S.;Zheng,Q.C.;Zhang,H.X.;Chu,H.Y.;Sun,C.C.Acta Phys.-Chim.Sin.2009,25,417.[赵勇山,郑清川,张红星,楚慧郢,孙家钟.物理化学学报,2009,25,417.]doi:10.3866/PKU.WHXB20090304
(19)Zhang,J.L.;Zheng,Q.C.;Zhang,H.X.Comput.Biol.Chem.2011,35,50.doi:10.1016/j.compbiolchem.2011.01.001
(20) Humphrey,W.;Dalke,A.;Schulten,K.J.Mol.Graph.1996,14,33.doi:10.1016/0263-7855(96)00018-5
(21) Darden,T.;York,D.;Pedersen,L.J.Chem.Phys.1993,98,10089.doi:10.1063/1.464397
(22) Hoover,W.G.Phys.Rev.A1985,31,1695.doi:10.1103/PhysRevA.31.1695
(23) Hénin,J.;Chipot,C.J.Chem.Phys.2004,121,2904.doi:10.1063/1.1773132
(24) Zhang,J.L.;Zheng,Q.C.;Zhang,H.X.J.Phys.Chem.B2010,114,7383.doi:10.1021/jp9113078
(25)Zhang,J.L.;Zheng,Q.C.;Li,Z.Q.;Zhang,H.X.PLoS ONE2013,8,e53811.
(26) Zhang,J.L.;Zheng,Q.C.;Zhang,H.X.Chem.Phys.Lett.2010,484,338.doi:10.1016/j.cplett.2009.12.022
猜你喜欢
生物化学与生物物理进展(2022年7期)2022-07-25
生物化学与生物物理进展(2022年6期)2022-07-21
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19
世界农药(2019年3期)2019-09-10
世界农药(2019年3期)2019-09-10
武警医学(2018年10期)2018-11-06
湖南农业(2016年12期)2016-03-10
现代农业(2016年4期)2016-02-28
天津科技大学学报(2015年2期)2015-08-09
火炸药学报(2014年3期)2014-03-20
