
利用连接介导PCR检测外源基因整合位点体系的建立
2013-09-20 13:25张冬杰
东北农业大学学报 2013年12期
刘 娣,李 斌,张冬杰
(1.黑龙江省农业科学院,哈尔滨 150086;2.东北农业大学动物科学技术学院,哈尔滨 150030)
外源基因整合位点检测,是鉴定转基因动物的最基本、最重要步骤。整合位点侧翼序列是外源基因表型研究和功能探讨的前提条件,对目的基因正常转录和表达有重要影响[1]。外源基因整合到宿主基因组发生在DNA复制S期,以多位点和单一位点形式随机插入受体基因组中的任意位置,存在毒性整合、有效整合和沉默整合三种整合状态,其表达水平不同[2]。目前检测外源基因整合位点方法主要通过分子生物学和细胞生物学方法进行检测,主要有PCR法,DNA斑点杂交,Southern印迹法等,其中以PCR方法最为简单、快捷。人们以PCR技术为基础设计了很多种扩增未知序列的染色体步移方法,如反向PCR法(inverse PCR,I-PCR),热不对称交错PCR法(thermal asymmetric interlaced PCR,TAIL-PCR)和连接介导PCR法(ligation mediated PCR,LM-PCR)[3-4]。
LM-PCR法的基本原理是首先将基因组DNA用特异性内切酶进行酶切,而后连接单链、双链或部分双链的接头,用序列特异引物和接头引物引发PCR扩增。通过对PCR扩增产物的克隆测序,即可获得外源基因的整合位点信息。但在PCR反应过程中,易发生单链或部分单链接头的凹入末端被补平,或接头的单链部分被降解等现象,这些副反应都将导致非特异扩增的增加,而难以得到特异性产物。为降低副反应的发生从而提高产物的特异性,TaKaRa和Clontech公司都通过对接头进行氨基修饰以抑制接头引物的扩增,接头的5'末端没有磷酸基,所以不能与酶切基因组的3'末端连接,形成缺口,这样就可以高效且特异性地扩增外源基因侧翼序列。目前,LM-PCR技术在外源基因定位方面应用较多,但是由于其扩增长度往往比较短(<1kb),若想获得更长的片段只能通过几次步移后才能得到[5-7]。
黑素皮质素受体4(MC4R)基因是影响猪生长肥育性能的主效基因之一,是参与调控体重、采食和能量平衡的关键信号物质[8-9]。本实验室在前期研究中获得转猪MC4R基因的转基因小鼠。为检测转入猪MC4R基因在小鼠基因组上整合位点,本研究采用LM-PCR法对外源基因的整合位点进行检测,以期建立转基因动物检测体系,为转基因动物研究提供理论依据。
1 材料与方法
1.1 材料
1.1.1 转猪MC4R基因小鼠DNA
黑龙江省农科院畜牧研究所分子实验室冻存。
1.1.2 工具酶,试剂盒及其他试剂
限制性内切酶、dNTP、DNA连接酶、DNA Marker、rTaq酶均购自Takara公司;西班牙琼脂糖、胰蛋白胨、二水乙二胺四乙酸二钠盐(EDTA-Na2·2H2O)、酵母提取物、三羟甲基氨基甲烷(Tris)均购自哈尔滨英俊生物技术有限公司;PCR纯化试剂盒购自北京天根生化公司。
1.2 方法
1.2.1 带有接头引物及特异性引物序列设计
参考Clontech公司试剂盒内接头序列合成本试验所用的接头,接头序列为:
5'GTAATACGACTCACTATAGGGCACGCGTG⁃GTCGACGGCCCGGGCTGGT 3'和5'PO4-ACCAG CCC-NH23';根据接头序列设计接头引物AP1和AP2,AP1:5'GTAATACGACTCACTATAG GGC 3',AP2:5'ACTATAGGGCACGCGTGGT 3';根据插入的基因片段启动子区序列(pcDNA3.1(+)PCMV区)设计特异性引物SP1和SP2,SP1:5'TAATA GGGTGGTGACAATGGTTTCTG 3';SP2:5'GT CG⁃GTCAAGCCTTGCCTTGTTGTAG 3',送交Invitrogen公司合成。
1.2.2 基因组的酶切与纯化
随机选取1只转基因阳性鼠的基因组为试验材料,分别用4种限制性内切酶(DraⅠ,EcoRV,PvuⅡ,SspⅠ)37℃过夜酶切。反应体系为:25 μL基因组 DNA(0.1 μg· μL-1),8 μL 限制性酶(10 units· μL-1),10 μL限制性酶缓冲液(10X),57 μL去离子水。酶切结束后,取5 μL酶切产物做琼脂糖凝胶电泳检测并观察结果。
采用苯酚-氯仿方法纯化酶切后的基因组DNA。①对每个反应管中加入等体积量(95 μL)的苯酚,低速离心,转移上层(水相)到一个新的1.5 mL EP管中。②在每个EP管中,加入等体积量(95 μL)的氯仿,低速离心,移上清到1个新的1.5 mL EP管中。③每个EP管加入2倍体积(190 μL)冰的 95%乙醇,1/10体积(9.5 μL)3 mol·L-1NaOAc(pH 4.5)和20 μg糖原,在4 ℃ 14 000 r·min-1下离心15 min。④倒出上清,用100 μL 80%的冰乙醇清洗涤沉淀,在4℃14 000 r·min-1下离心10 min,室温下干燥沉淀,加入20 μL TE(Tris:EDTA=10:0.1,pH 7.5)溶解沉淀,并从每个管中取出1 μL溶液在0.5%琼脂糖中电泳检测。
1.2.3 纯化后的基因组DNA连接接头
连接体系为:4 μL纯化后的基因组DNA,1.9 μL接头(25 μmol· L-1,1.6 μL 10X ligation buffer,0.5 μL T4DNA连接酶(6 units·μL-1)。16℃过夜连接。
1.2.4 两轮PCR扩增检测
将上一步连接产物加入72 μL TE(Tris:EDTA=10∶1,pH 7.5)稀释并在70℃下5 min终止反应。以此为PCR模板,以AP1和SP1为引物进行第1轮PCR扩增,反应体系:40 μL去离子水,5 μL Buf⁃fer,1 μL dNTP,1 μL AP1,1 μL SP1,1 μL酶切产物,1 μL LA Taq酶,下同。反应条件为:7 cycles:94℃ 25 s,72℃ 3 min;32 cycles:94℃25 s,67℃3 min;67℃7 min。PCR反应结束后,取5 μL PCR产物做琼脂糖凝胶电泳并分析结果。对扩增出条带的产物,用去离子水50倍稀释作为第2次PCR扩增模板,以AP2和SP2为引物做第2轮PCR扩增,反应条件为:5 cycles:94℃25 s,72℃ 3 min;20 cycles:94℃25 s,67℃ 3 min;67℃7 min。取5 μL酶切产物做琼脂糖凝胶电泳并分析结果。
1.2.5 克隆测序与序列比对
将第2轮PCR扩增产物的目的条带回收纯化,连入pMD18-T载体,转化入大肠杆菌后,摇菌,送交Invitrogen公司测序。将测得的结果采用BLAST方法与NCBI网站上已提交的小鼠基因组序列进行比对。
2 结果与分析
2.1 基因组的酶切与纯化
转基因阳性小鼠的基因组DNA经EcoRV,PvuⅡ,DraⅠ,SspⅠ过夜酶切后,均呈现出弥散状态(见图1),说明基因组DNA已经被切开,可用于后续试验。
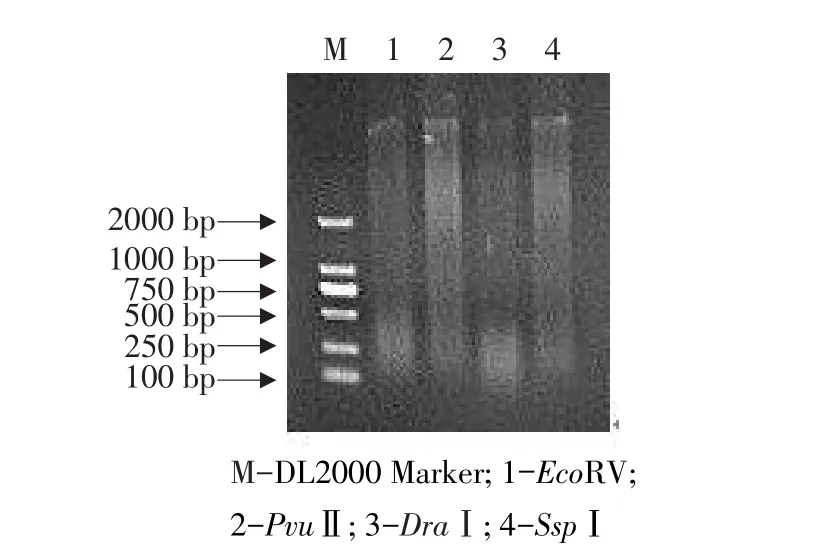
图1 基因组酶切Fig.1 Enzyme digestion of genome
2.2 两轮PCR扩增检测结果
基因组分别经酶切后,第1轮PCR反应后,EcoRV和PvuⅡ酶切后的基因组没有扩增出特异条带,DraⅠ和SspⅠ酶切后的基因组扩增出特异条带(见图2)。以EcoRV,PvuⅡ,DraⅠ和SspⅠ分别对应的第1轮PCR产物为模板进行第2轮PCR扩增,仍旧是DraⅠ和SspⅠ所对应的产物获得了单一的特异条带,大小在250~1 000 bp(见图3)。

图2 第1轮PCR扩增结果Fig.2 First PCR amplification results

图3 第二轮PCR扩增结果Fig.3 Second PCR amplification results
2.3 克隆测序与序列比对
对上述试验所获得的2条特异条带克隆测序,经过序列比对发现,2条特异条带实为同一个序列片段(见图4),只是片段大小不同。以其中的长片段为目的片段,通过序列比对发现,其中133 bp为已知的载体序列,430 bp为未知序列(见图5)。以该序列为源序列,与NCBI网站已提交的小鼠基因组序列比对后发现,所克隆到的未知序列与小鼠6号染色体37 236 325~37 236 754之间的序列100%同源(见图6)。

图4 两条特异条带克隆序列比对结果Fig.4 Result of Two specific bands cloned sequence blast
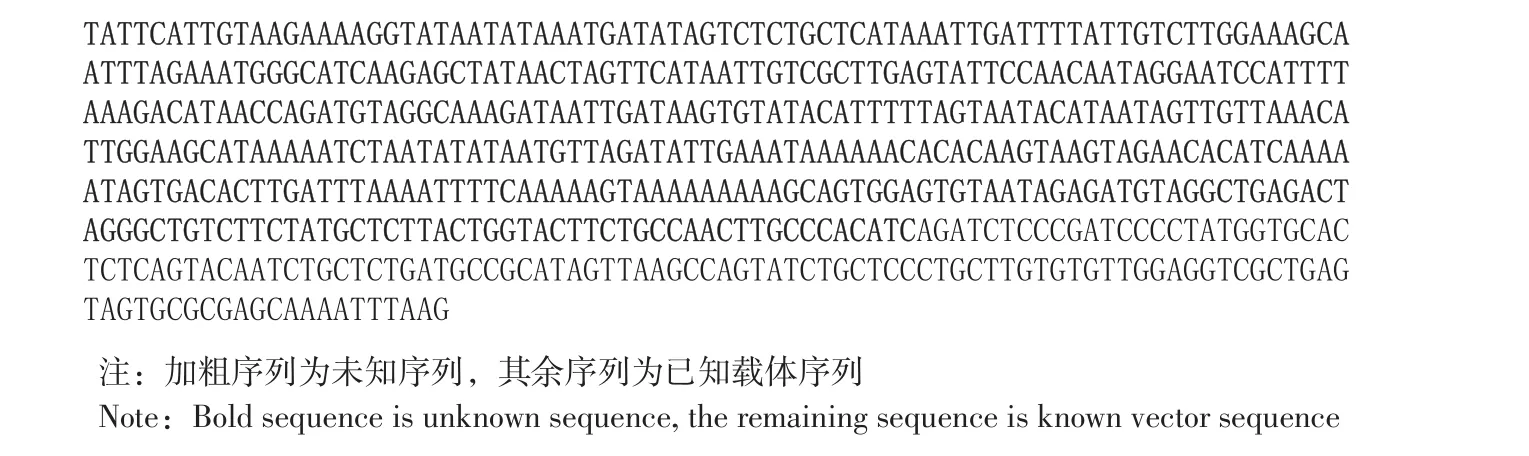
图5 序列信息Fig.5 Sequence information
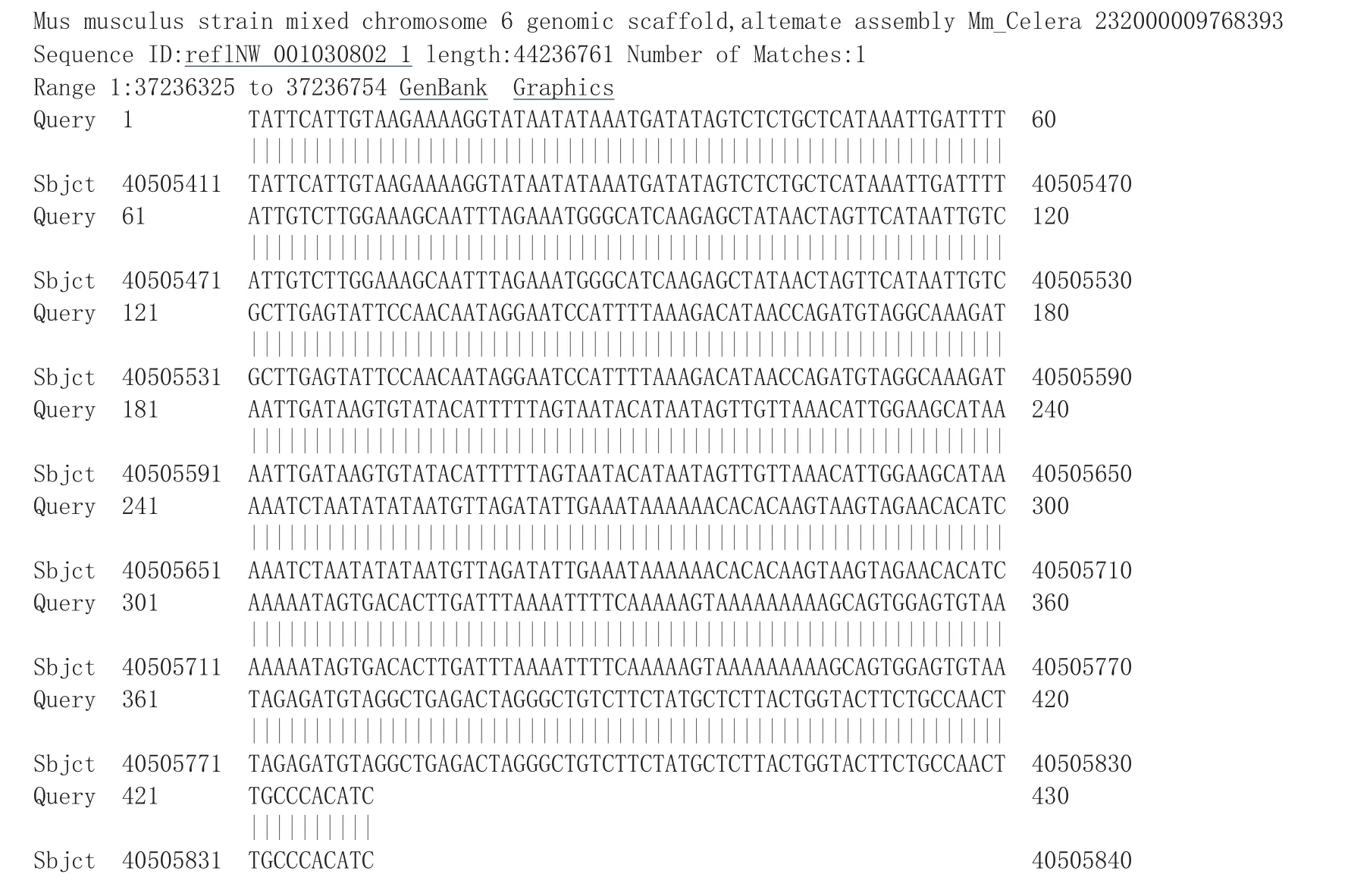
图6 Blast比对结果Fig.6 Blast result
3 讨论与结论
外源基因的整合受多因素影响,如DNA构型,DNA的浓度和进入宿主细胞的方式等。一般认为,外源基因整合位点对其表达水平有重要影响,同样一个外源基因可在一定的整合位点正常表达,而在另一些位点不表达或低水平表达。所以,外源基因整合位点的检测对研究外源基因的表型和功能十分重要[10]。
目前扩增外源基因整合位点方法很多,如LM-PCR,TALL-PCR等。本试验通过LM-PCR测得外源基因整合位点,对接头的3'端采用氨 基酸修饰克服单引物扩增,并提高扩增效率[11]。本试验只得到一个整合位点(Blast比对后发现2条特异条带为同一整合位点),可看出LM-PCR方法 虽然结果准确但效率较低,操作比较复杂(需要一系列处理,如酶切,连接纯化),扩增片段较短(<2 000 bp)。可能会有更多整合位点,但本试验仅检测出一个,其余整合位点还需通过更换内切酶的方法进一步检测。在试验过程中发现,第一轮PCR扩增很难扩增出特异条带,甚至无特异条带,但是如果将稀释倍数放小,在第二轮PCR扩增时也会扩增出特异条带,可能是限制性内切酶酶切位点距离目的基因过远或接头序列与酶切产物的连接效率较低,导致PCR很难扩增出目的片段。如果能克服酶切的不确定性,将使LM-PCR的染色体步移方法成功率提高。由此可见,使用LM-PCR进行外源基因整合位点检测虽然有效,但反应参数仍需完善。
本研究采用LM-PCR方法检测到整合到小鼠基因组上的猪MC4R基因,该整合位点位于小鼠第6号染色体的37 236 754 bp处。初步建立了采用LM-PCR对外源基因整合位点检测的体系,可为今后转基因动物的检测奠定基础。
[1]吴波,朱作言.转基因动物整合位点的研究进展[J].遗传,2003,25(1):77-80.
[2]谭贝贝,苏红,胡嘉祺.转基因克隆牛外源基因整合位点的研究[J].中国农学通报,2012,28(17):28-32.
[3]孔庆然,武美玲,朱江,等.转基因猪中外源基因拷贝数和整合位点的研究[J].生物化学与生物物理进展,2009,36(12):1617-1625.
[4]Liu Y G,Whittier R F.Thermal asym metric interlaced PCR:auto⁃matable amplification and sequencing of insert end fragmentsfrom P1 and YAC clones for chromosome walking[M].Genomics,1995,25(801):674-681.
[5]王闵霞,马欣荣,王天山,等.染色体步行PCR技术[J].应用与环境生物报,2006,12(3):427-430.
[6]Redstone JS,WoodwardMJ.The developmentofa ligasemediated PCR with potential for the of serovars with Leptospira interrogans[J].Veter Microbiol,1996,51(3):351-362.
[7]Tormanen VT,Seiderski PM,Kaplan BE,et al.Extension product capture improves genomic sequencing and DNaseÑ footprinting by ligation mediated PCR[J].Nucleic Acids Research,1992,20:5487.
[8]霍明东,王守志,李辉.MC4R基因多态性与鸡生长和体组成性状的相关研究[J].东北农业大学学报,2006,37(2):184-189.
[9]宋军,王振坤,孔庆然,等.利用体细胞核移植技术生产表达绿色荧光蛋白转基因猪胚胎的研究[J].东北农业大学学报,2008,39(3):64-69.
[10]王继英,杜立新.转基因动物制作及提高外源基因表达的策略[J].中国畜牧兽医,2002,2(92):30-34.
[11]刘春香,张卫华,孙小镭.基于PCR的染色体步移中单引物扩增的克服与非特异引物的利用[J].中国生物化学与分子生物学报,2010,26(4):380-385.
猜你喜欢
学与玩(2022年10期)2022-11-23
舰船科学技术(2022年11期)2022-07-15
今日农业(2022年3期)2022-06-05
西藏农业科技(2019年3期)2019-11-04
山西地震(2019年1期)2019-03-20
西安交通大学学报(2019年3期)2019-03-08
现代园艺(2018年3期)2018-02-10
上海农业学报(2017年3期)2017-04-10
系统工程与电子技术(2016年2期)2016-04-16
创新科技(2015年1期)2015-12-24
