
基于粒子系统的大分子三维常规表达可视化
2013-09-25 02:58吕智涵苏天赟
图学学报 2013年4期
吕智涵, 苏天赟
(1. 中国科学院深圳先进技术研究院,广东 深圳 518055;2. 国家海洋局第一海洋研究所,山东 青岛 266061)
基于粒子系统的大分子三维常规表达可视化
吕智涵1, 苏天赟2
(1. 中国科学院深圳先进技术研究院,广东 深圳 518055;2. 国家海洋局第一海洋研究所,山东 青岛 266061)
随着众包成为大数据问题的流行解决方案,虚拟社区逐渐被应用到科学研究中,并借助手机交互,人们在追求高质量渲染的同时也开始探索渲染方法的普适性。本文研究一种蛋白质大分子三维可视化方法,该方法通过粒子系统管理大量原子,改进着色方法减少每个原子渲染时间,可以表达甘草和范德华模型,二级结构等常规表达。选取从 103至105尺度的分子作为用例,与流行方法比较静止和交互时的效率,均具有更高效率,更适于移动平台。对分子结构进行域分解优化,进一步提高交互性能。成果发布至手机,可流畅运行。
分子可视化;虚拟科学社区;虚拟世界;大数据;粒子系统;常规表达;OpenGL es2.0
目前国内外生物学领域的研究迫切需要对大分子结构,组织网络或者甚至器官和组织体[1-3]等庞大而复杂的系统进行可视化的工具。
以平面图片表达分子结构的可视化方式容易引起误差[4],而新的基于虚拟现实技术的可视化方法使得科学家能够有效的与分子结构互动。虚拟现实技术可以解决分享、组织和注释分子结构可视化问题,并促进科学家间高效协作。高性能协同虚拟环境提供了解决方案[5],但推广应用却是挑战。在大数据(Bigdata)挑战的背景下,众包(Crowdsourcing)逐渐流行,并已表现出实用价值[6]。该方法在参与科学,生物学和计算机科学相结合的虚拟科学社区领域广受关注,有可能因此取得巨大进展[7,8]。
虚拟科学社区可以作为一个教育平台[9]和社会,行为,经济等学术领域的研究平台[10]。此外,科学的项目启动源于普通民众的调查[11],虚拟科学社区可以完成特定的调查任务,并用来解决科学问题,最近被用来预测蛋白质折叠[12]并产生显著成效[13]。随着手机等轻量级设备计算能力的增强,手机设备上的虚拟科学社区逐渐显现出巨大潜力。对于生命科学的社区,瓶颈之一是蛋白质大分子的三维可视化技术。
蛋白质大分子三维可视化方法发展的主要目标,就是通过改进现存的渲染技术去克服巨大结构等表达限制。有很多不同类型的分子表达存在,分子表达从最简单的空间填充和范德华球到分子表面表达定义更复杂的大分子的整体形状[14]。在这两个极端情况下,可以定义常规分子表达如球棒,甘草或者范德华表达,或者生物聚合物的主键结构。其中最流行的是基于线(例如球棍和甘草)和基于色带(二级结构)的模型,“因为他们以简单的可视化形式呈现出每个分子的重要结构属性”[15]。本文方法对球棍,甘草和范德华模型,二级结构分别进行表达。
1 研究现状
最古老常用的结构表达例如球棒,范德华(也称空间填充或Cpk)或甘草等简单的分子表达属于常规表达,首先通过简单的线去连接原子(简化甘草模型)或者基于更复杂的圆柱和球的模型(球棒,空间填充和甘草模型)。迄今为止甘草表达一般被三角球面和圆柱形表面实现。该方法的主要局限性在于渲染巨大的集合时要创建大量的三角面,例如,创建一个平滑的球,至少要大约300个三角面,因为包括视野外的所有三角面都渲染,所以影响了性能。即使用最新图形硬件渲染如此庞大的三角面数量速度也会有延迟。
随着可编程图形卡的开发,诞生了运行于GPU的光线投射/光线追踪新技术。首先创建一个粗略的三角信封,然后用可编程图形管道精确的定义解析表面。采用这种方法,可以实时的表达面向任何缩放级别的像素精确精度的大量的球和圆柱体。
GPU 光线投射首次用于分子表达由 Toledo和Levy在2004年实现,该方法已在VMD分子浏览器中实现[16]。与此同时,Bajaj等的TexMol方案也使用基于GPU技术[17]的二级结构表达。2006年,这种方法被Sigg等[18]和Tarini等[19]扩展,以表达带有特殊光效的巨大分子并强化图形预测能力。2007年,Lampe等使用面向空间填充或球棒模型的 GPU光线投射技术表达一个二级结构来可视化大分子的动态变化[20]。这些例子基于 GPU的表现改善了实时性和可交互操作。D.S.Goodsell描述了几种技术使得分子可以向细胞尺度可视化过渡[21]。Chavent开发了HyperBalls分子表达方法[22],实现高质量的Raycasting,用立方体和平行六面体分别生成信封进而渲染为原子和键,以双曲面代替圆柱体来表达键连接球,可以表达分子键的渐进演变。
浙江大学CAD&CG国家重点实验室对蛋白质分子三维数据场的特征抽取和可视化进行研究[23],成功从数据场中抽取蛋白质进行分析[24],并成功地观察到HIV-1蛋白酶分子中隐藏的水分子排出通道[25]。厦门大学固体表面物理化学国家重点实验室[26]在网页上对三维分子进行了实现并用于网络教学。
随着图形卡技术的快速发展分子表达逐渐细化品质提升,而针对手机显卡等轻量级设备的渲染技术却并没有随之发展。以HyperBalls为例,尽管其独特动态效果可以逼真的模拟分子动态效果,但是依赖于昂贵的 GPU显卡,曾以iPhone4s和 HTX one作为样机分别测试了 IOS系统和android系统下的OpenGL ES2.0框架下的HyperBall实现,均无法直接支持效果,且运行效率极低。
本文所介绍的方法以降低光照实时效果为代价,以粒子系统管理大量信封,实现适用于手机等普适设备。
2 粒子系统表示球棒模型
对 PDB文件进行分析以提取原子信息,包括原子类型,残留物名称,残留ID和坐标。分子的拓扑结构由原子间的距离创造键的算法构建。
2.1 点精灵粒子系统创建
网格图形以大量三角面表达平滑球,因此不适合实时渲染分子。本文使用一个总是面向垂直屏幕的平面的正方形(仅由两个三角组成)表达单一的原子。将一个图像粘贴到这个正方形内,称作点精灵,也称广告牌 (Billboard)。这个方法可以有效的显示很多相似的图像。在虚拟世界中,该方法通常用来描述涉及很多小粒子的虚拟效果,例如:火,烟,云,雪,沙,等。这些粒子的总和称为粒子系统。在每个粒子系统正方形粒子中粘贴球形图像可以表达原子,如图1所示。
图 1左为球形表示原子点精灵创建过程示意。步骤A创建朝向垂直于屏幕的方向的正方形作为信封。步骤 B在正方形平面上粘贴球形图像。步骤C表示了3D点精灵在屏幕上的可视化。对于步骤 A、C,P 表示屏幕平面;左边是屏幕上的结果,右边是3D空间中的图形。其中步骤B中的球形粘贴过程由改进的Alpha混合着色器算法实现。由于蛋白质分子表达的默认使用固定的几种颜色抽象表达有限的原子类型,比如红色代表氧,绿色代表碳,所以,直接对RGB 3个通道的值进行赋值,以此替换原算法中以灰度与颜色矩阵相乘的过程,使时间复杂度由O(n^3)降低到O(n)。实践中由于多次使用if语句,且颜色矩阵中n仅为3,所以在测量小分子时,效率并未有显著提升,而当分子尺寸提升到5万以上时,效率较大提升。着色步骤如下:
1) 选择纹理源为绿色静态 Hyperballs截图,RGB值为(Ro, Go, Bo),其中 Ro=0,0<Go<=255, Bo=0,如步骤B中左侧绿色球形图像;
2) 判定目标颜色,逐像素对 RGB通道分别赋值,如表1所示。

表1 RGB替换表
图1右上为以线段表示键点精灵的创建。两球半径分别为R1和R2,球膨胀系数为S1,键膨胀系数为S2,则键点精灵的为两底宽为D1=R1*S1*S2 和D2=R1*S1*S2 (0<S1<=2, 0<S2<=2)的等腰矩形,矩形高H为两原子圆心的空间距离。颜色为由球1到球2的渐变色,由Alpha混合着色器生成。当S1=0.2,S2=2的时候为甘草模型。当S1=0.2,S2=2的时候为甘草模型。当S1=1,S2=0.4的时候为球棒模型。当S1=1,S2=0的时候为范德华模型。
图 1右下为以粒子系统渲染的 PDBID为3AFA的分子的球棒模型,原子数 12003,键数12809。小窗口内为分子的局部细节。
2.2 粒子系统与其他方法性能对比
对比试验分为3步:(1) 选取简单分子作为测试用例评估不同的方法;(2) 进一步选择一系列原子数从103到105的分子结构作为测试案例,以选择最有效的方法;(3) 以匀速旋转模拟交互移动时的速率测试。
测试用机为 Mac Pro,配置为双核 Intel Xeon 3 GHz 处理器,8 GB内存,NVIDIA GTX 285显卡,操作系统64位Mac OS X 10.6。
测试1 以分子ferrocytochrome C, PDB ID:1KX2作为测试用例对比几种实现方法。该蛋白质由1249个原子和1113个键组成,是普通尺寸的PDB文件。
我们对4个渲染原子的方法评估:粒子系统、三角面球、三角面立方体和HyperBalls球。我们也测试 3种显示键的方法:立方体、线段和HyperBoloids双曲面。如图2所示,表示3D粒子系统是显示球的方法中最高效的。其次是三角立方体,比 3D粒子系统慢 8倍,之后是HyperBalls实现的球和三角面球。
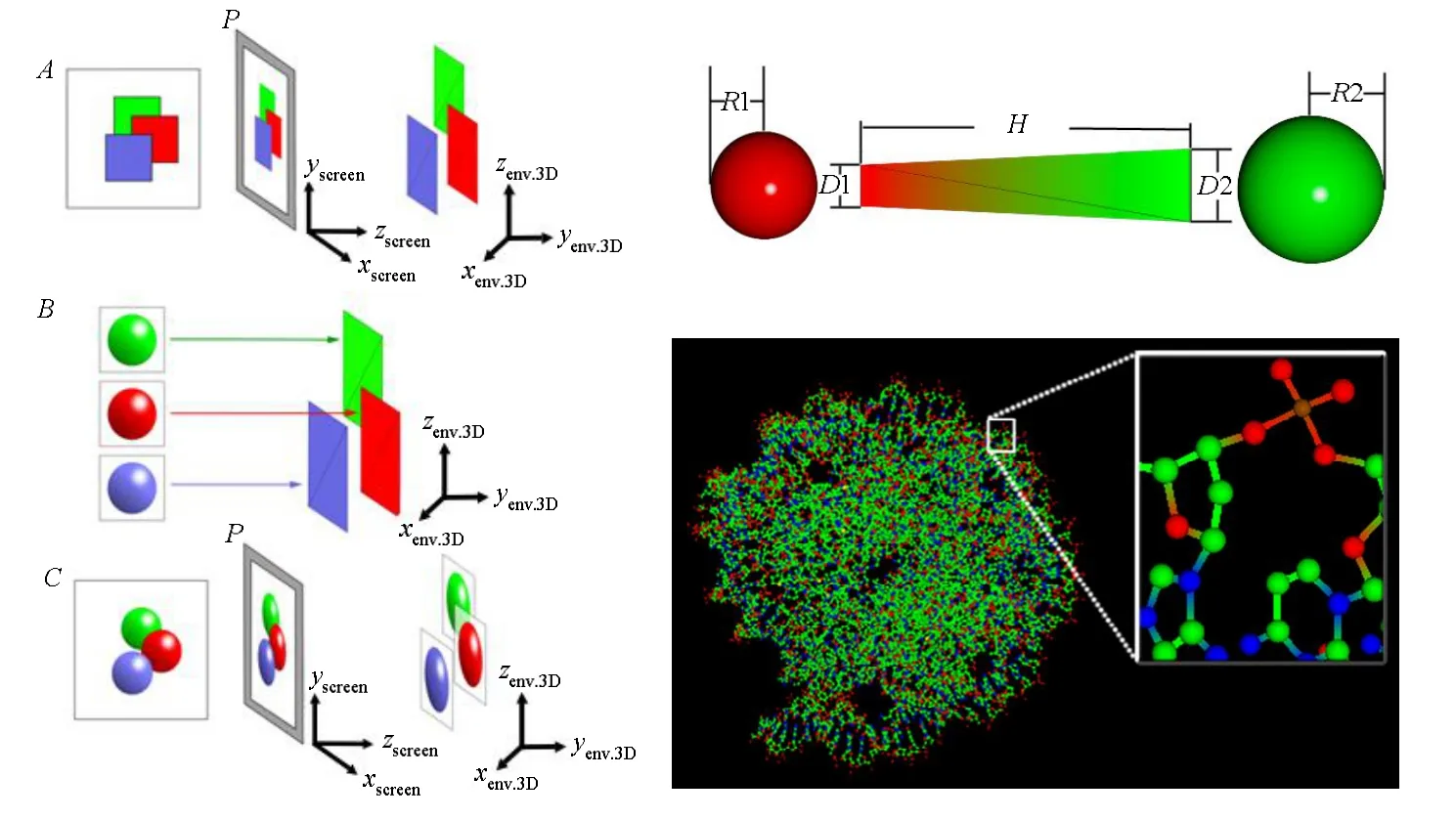
图1 原子与键的点精灵粒子系统创建
在原子上附加渲染键并没有改变这个排序,但是,可以观察到显示性能显著下降。对于粒子系统,显示刷新率下降了5倍,而对于其他方法下降近2倍。当使用HyperBoloids或线来渲染键时该值对于粒子系统减少了超过 10倍。对于HyperBalls和三角面球表达,加入线或HyperBoloids将显示速度减少3倍。对于粒子系统的巨幅变化可以解释为其原本高效率渲染球,而对键的渲染添加了其他的图形方法限制了效率。其他方法的性能变化是由于当键添加的时候顶点数量接近2倍。在用于渲染键的图形基元中,线段是最高效的,立方体和双曲面键的性能略低。
对于实时交互操作,若FPS低于10帧则会有交互延迟。图2表示添加键后的所有方法的运行性能以及它们间的细微差别。根据所选方法不同,图形基元的三角型和顶点的数目会有显著变化,并直接影响显示效率。绘制这些元素的函数的调用次数是另一个重要的瓶颈,调用越少效率越高(表2分别对几种方法渲染分子的三角面,定点数目和绘制次数进行统计)。总体的渲染性能取决于渲染窗口大小和内置图形硬件性能。
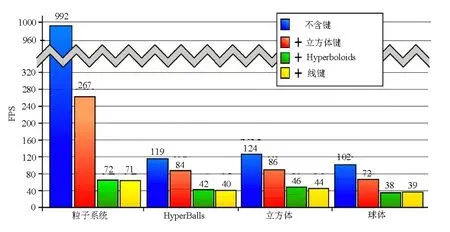
图2 性能对比
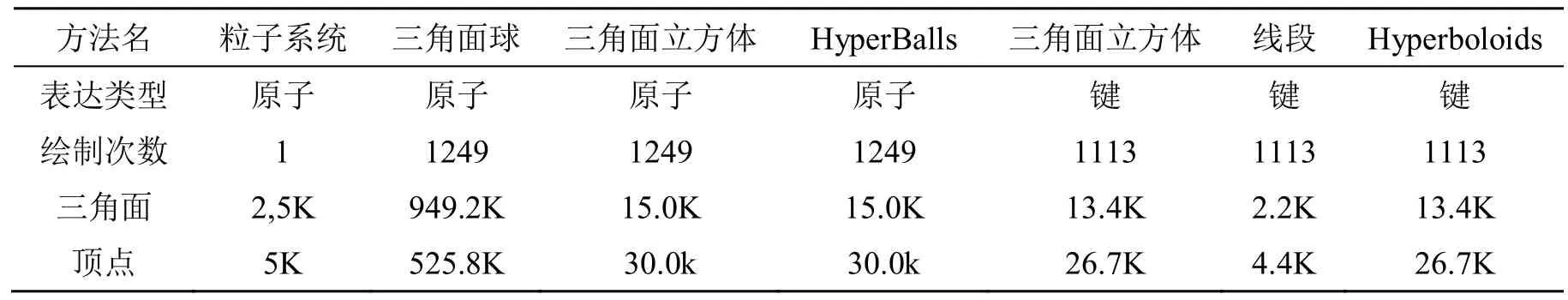
表2 几种方法渲染1×k2分子的三角面,顶点的数目和绘制次数
测试 2 选择粒子系统,三角面球和HyperBalls方法,以一个尺寸从1200到100000个原子的分子集来评估他们的效率限制,如表3所示(表3为测试所采用的分子集合)。这个实验以球形原子表达巨大分子结构,目的是渲染最大数量的原子来评估这些方法的真实性能。如图3所示,首先评估静态分子的显示率,见图3左。粒子系统的性能显然优于另外两种方法。甚至对于由10万个原子组成的ATP合酶(PDBID为3OAA)等超大分子,帧速率仍然很高。HyperBalls球的结果是从几个分子到几千个原子的分子:超过10000个原子时FPS值接近10帧。三角面球渲染效率略低于 HyperBalls球,可流畅显示约6000个原子。
测试3 通过对粒子系统球测量当分子被操作时的 FPS值分析了用户交互对于显示性能的影响。为仿真用户操作,以固定速率自动旋转分子来模拟用户移动,结果见图3右。旋转式移动对于HyperBalls球,立方体或者三角面球的性能基本没有影响,而对于粒子系统有显著的影响。随分子变大影响更明显,这可以解释为所有粒子的深度排序更加耗时。

表3 评估性能所用的一系列分子
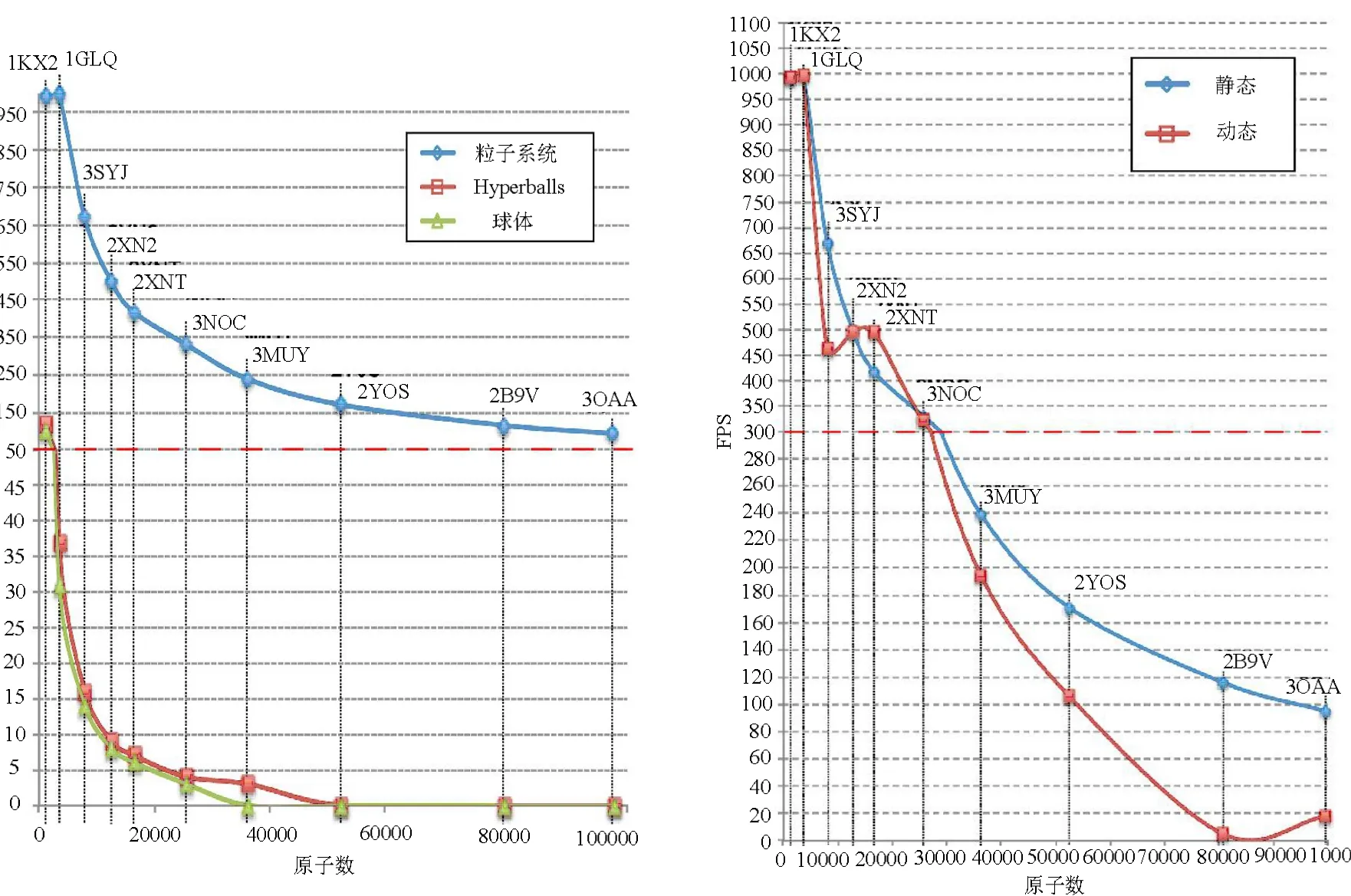
图3 测试效率限制
为拟补该问题,将分子结构分解为 64个空间块来实现域分解,如图4所示。每块由一个独立的粒子系统表示。域之间以坐标轴方向排序,且以每个域为对象进行视锥筛选和遮挡剔除。该方法为大分子改进效率并且让用户可以交互操作50000个原子的分子。若不进行域分解,则交互渲染超过 15000个原子将会引起速度显著下降。
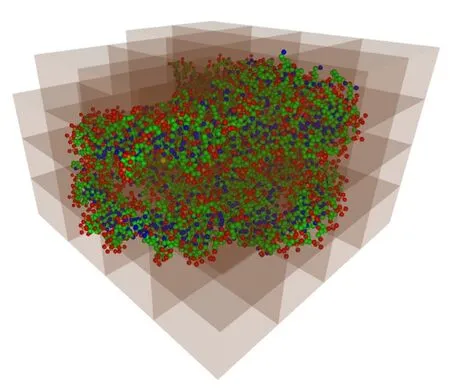
图4 域分解
2.3 粒子系统表示二级结构
蛋白质分子的二级结构只选取α-碳原子,如图 5所示。对所选 α-碳原子得坐标以 Hamilton差值计算出平滑的坐标集,分别以粒子系统和Hyperballs方法渲染。选取测试用例1kx1,由7130个原子,7033个键组成。以粒子系统实现的帧率为 65FPS,以 Hyperball+Hyperstick实现的帧率为8FPS。
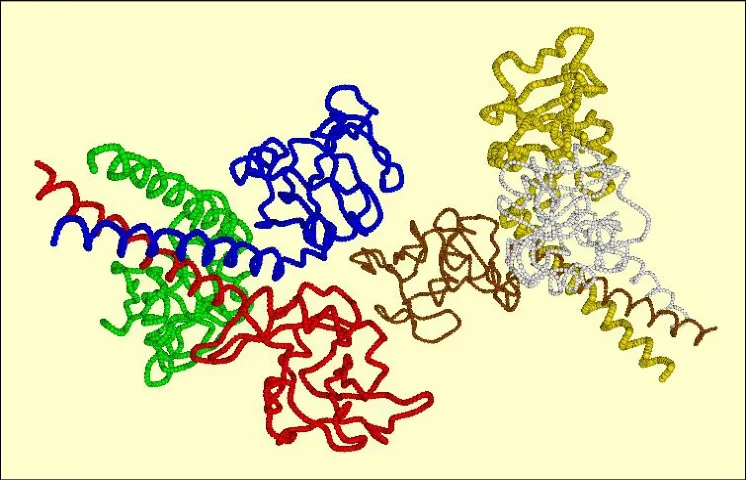
图5 二级结构
2.4 粒子系统表示静电势
静电势可视化通常表示为等值面或是沿着磁力线的颗粒动画,如图6所示。静电势格网由APBS[27]计算得到的静电电位网格梯度矢量场的粒子平流计算得到,然后生成 OpenDX格式文件,并可以为等值面可视化读取,从BioBlender[28]预处理生成描述了每个磁力线的动画的点坐标的JSON文件来表达磁力线。
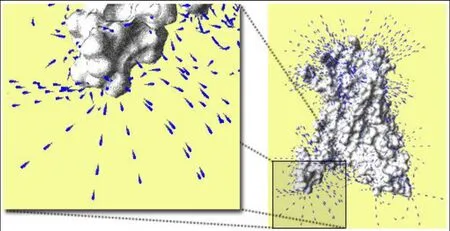
图6 静电势
磁力线坐标加载到内存中后,通过Hamilton差值计算出平滑曲线坐标,使用粒子系统重建静电势,并着色器渲染一条描绘粒子运动的虚线,这个着色器根据一个定时器来实现沿着磁场线的正弦函数的动画。表示静电势颗粒的粒子的颜色,宽度,长度和速度可以更改。
分子表面可以从Chimera[29],VMD[16]等分子建模程序导出为obj格式的三维三角网格。面重建后通过着色器对表面渲染简单光照和均匀颜色来突出立体感,以衬托静电势路径。
2.5 手机上的渲染
在iPhone3GS上分别实现了基于粒子系统和Hyperballs的三维分子。基于Hyperballs的实现运行效率很低,用户交互延迟很明显,没有实用价值,而且需要对OpenGL ES2.0的支持;而基于粒子系统的实现运行流畅。
3 结 束 语
本文研究一种适用于手机等轻量级交互设备的蛋白质大分子渲染方法,该法基于粒子系统,可以表达球棒,甘草,范德华模型,二级结构,静电势。经测试该法运行效率更高,更适合手机等计算性能较低,屏幕较小对显示质量要求略低的设备。这种方法将为以虚拟科学社区为主要交流平台的众包提供面向手机设备的基础可视化方案。
致谢 感谢法国国家科学研究中心UPR9080实验室主任Marc Baaden博士为本文提供的指导意见和对项目顺利实施给予的帮助。
[1]O'Donoghue S I, Goodsell D S, Frangakis A S, et al.Visualization of macromolecular structures [J]. Nat Methods, 2010, 7:42-55.
[2]Gehlenborg N, O'Donoghue S I, Baliga N S, et al.Visualization of omics data for systems biology [J].Nat Methods, 2010, 7:56-68.
[3]Walter T, Shattuck D W, Baldock R, et al. Visualization of image data from cells to organisms [J]. Nat Methods,2010, 7:26-41.
[4]Ottino J M. Is a picture worth 1,000 words [J]. Nature,2003, 421:474-476.
[5]Kolatkar A, Kenndey K, Halabuk D, et al. C-ME:a 3D community-based, real-time collaboration tool for scientific research and training [J]. PLoS One 2008:3.
[6]Sansom C. The power of many [J]. Nat Biotechnol,2011, 29:201-203.
[7]Kawrykow A, Roumanis G, Kam A, et al. Phylo:a citizen science approach for improving multiple sequence alignment [J]. PLoS ONE, 2012, 7:31-32.
[8]Eiben C B, Siegel J B, Bale J B, et al. Increased Diels-Alderase activity through backbone remodeling guided by Foldit players [J]. Nat Biotechnol, 2012, 30:190-192.
[9]Mayo M J, Video games:a route to large-scale STEM education [J]. Science, 2009, 323:79-82.
[10]Bainbridge W S. The scientific research potential of virtual worlds [J]. Science, 2007, 317:472-476.
[11]Bonetta L. New citizens for the life sciences [J]. Cell,2009, 138:1043-1045.
[12]Cooper S, Khatib F, Treuille A, et al. Predicting protein structures with a multiplayer online game [J].Nature, 2010, 466:756-760.
[13]Khatib F, DiMaio F, Foldit C G, et al. Crystal structure of a monomeric retroviral protease solved by protein folding game players [J]. Nat Struct Mol Biol, 2011, 18:1175-1177.
[14]Edelsbrunner H. Discrete & computational geometry [J].1999, 21(1), 87-115.
[15]Goodsell D S. Structure [J]. 2005, 13(3):347-354.
[16]Humphrey W, Dalke A, Schulten K. VMD:visual molecular dynamics [J]. J Mol Graph, 1996, 14:33-38, 27-38.
[17]Bajaj C, Djeu P, Siddavanahalli V, et al. TexMol:Interactive visual exploration of large flexible multi-component molecular complexes [C]//Proceedings of the conference on Visualization '04,2004:243-250.
[18]Sigg C, Weyrich T, Botsch M, et al. 'GPU-based ray-casting of quadratic surfaces' [C]//Eds.,Eurographics Symposium on Point-Based Graphics.
[19]Tarini M, Cignoni P, Montani C. Ambient occlusion and edge cueing to enhance real time molecular visualization [J]. IEEE Trans Vis Comput Graph,2006, 12:1237-1244.
[20]Daae L O, Viola I, Reuter N, et al. Two-level approach to efficient visualization of protein dynamics [J]. IEEE Trans Vis Comput Graph, 2007,13:1616-1623.
[21]Goodsell D S. Visual methods from atoms to cells [J].Structure, 2005, 13:347-354.
[22]Chavent M, Vanel A, Tek A, et al. GPU-accelerated atom and dynamic bond visualization using hyperballs:a unified algorithm for balls, sticks, and hyperboloids [J]. J Comput Chem, 2011, 32:2924-2935.
[23]汪 莉, 万华根, 彭群生. 基于分子力学的蛋白质分子场可视化[C]//中国计算机图形学进展2008-第七届中国计算机图形学大会论文集. 2008.
[24]张 涛, 陈 为, 谢利广, 等. 基于三维数据场拓扑抽取的蛋白质结构分析[J]. 软件学报, 2006, 17:120-125.
[25]韩 玮, 汪 莉, 陈 为, 等. 面向大分子的三维数据场特征分析与可视化[J]. 软件学报, 2006, 17:103-109.
[26]陈明旦, 刘建文, 焦毓才, 等. Web 上的三维分子结构模拟[J]. 厦门大学学报(自然科学版). 2003,42(2):175-179.
[27]Baker N A. Electrostatics of Nanosystems:application to microtubules and the ribosome [J]. Proceedings of the National Academy of Sciences, 2001, 98:10037-10041.
[28]Andrei R, Callieri M, Zini M, et al. Intuitive representation of surface properties of biomolecules using BioBlender [J]. BMC Bioinformatics ,2012, 13(4):16.
[29]Pettersen E, Goddard T, Huang C. UCSF Chimera—a visualization system for exploratory research and analysis [J]. Journal of computational chemistry,2004, 25(13):1605-12.
3D Visualization of Macromolecular Conventional Expression Based on Particle System
Lü Zhihan1, Su Tianyun2
( 1. Chinese Academy of Science(CAS) Shenzhen Institutes of Advanced Technology, Shenzhen Guangdong 518055, China;2. The First Institute of Oceanography, SOA, Qingdao Shandong 266061, China )
The crowd sourcing is becoming a popular solution to big data problems, and virtual community is gradually applied to scientific research. People also begin to pursue the high-quality rendering as well the universal rendering methods. This paper studies a protein macromolecules 3D visualization method, which is to manage the large number of atoms by the particle system, so as to reduce the rendering time of each atom by improving the coloration method, thus it can express Licorice and Van der Waals model, the secondary structure and some other conventional expressions.Taking molecules from the 103to105scale as application examples, comparison of the static and interactive efficiency between the particle system method and the popular method shows that the former has better performances in both aspects. The domain decomposition of the molecular structure is optimized, and the interactive performance is improved apparently. The results can run smoothly when being published to the iPhone
molecular visualization; virtual science community; virtual world; big data;particle system; conventional expression; OpenGL es2.0
TP 391
A
2095-302X (2013)04-0119-07
2013-01-09;定稿日期:2013-01-30
国家自然面上项目,城市增强现实及搜索应用关键技术(61070147);文化部科技创新项目,基于增强现实的文物多维展示 系统平台研究(16-2012);深圳市科创委基础研究计划,基于格子玻尔兹曼方法的多视点建模技术研究(JC201105190951A)
吕智涵(1984-),男,山东青岛人,博士,助理研究员,主要研究方向为普适虚拟现实,普适增强现实。E-mail:lvzhihan@gmail.com
苏天赟(1977-),男,山东青岛人,博士,副研究员,主要研究方向为维空间数据管理和多维可视化。E-mail:sutiany@fio.org.cn
猜你喜欢
世界科学技术-中医药现代化(2022年3期)2022-08-22
师道·教研(2022年1期)2022-03-12
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
农村青少年科学探究(2020年5期)2020-08-18
海洋信息技术与应用(2020年1期)2020-06-11
传媒评论(2019年4期)2019-07-13
新民周刊(2018年8期)2018-03-02
饮食科学(2017年12期)2018-01-02
