
固相萃取/超高效液相色谱-电喷雾串联质谱法检测酶制剂中3-硝基丙酸
2013-10-08 00:51乐爱山孔祥虹
分析测试学报 2013年6期
张 璐,乐爱山,郑 玲,孔祥虹
(1.陕西出入境检验检疫局,陕西 西安 710068;2.广西出入境检验检疫局,广西 南宁 530022)
酶制剂是果汁加工过程中广泛使用的加工助剂[1-3],常用的有淀粉酶、果胶酶、蛋白酶、糖化酶、脂肪酶、超滤酶等。果汁生产中使用多种酶制剂以提高加工效率,增加产品稳定性,如使用淀粉酶来转化未成熟果实中的淀粉,使用果胶酶以除去榨汁中的果胶,使用超滤酶增加果汁超滤时的通过率。但用米曲霉发酵产生的酶制剂可能含有强毒物质3-硝基丙酸(3-Nitropropionic acid,3-NPA)。3-硝基丙酸是一种无色针状晶体,为高等植物受霉菌污染产生的有毒物质,可引起牲畜中毒,其中毒症状主要表现为中枢神经系统的损害[4-7],急性期表现有呕吐、眩晕、阵发性抽搐、昏迷等,严重可导致死亡。联合围粮农组织和世界卫生组织关于食品添加剂的联合专家委员会(Joint FAO/WHO Expert Committee on Food Additives)在1987即明确规定用米曲霉发酵生产的淀粉酶、蛋白酶、葡萄糖化酶、脂肪酶等时均要检测3-NPA。
目前对于3-NPA的检测主要集中在甘蔗及甘蔗制品[8-9],且检测方法多为高效液相色谱法[10]、气相色谱法[11]、薄层色谱法[8]等。但这些方法均存在样品前处理繁琐、需使用大量毒性大的有机试剂、干扰较大或需要进行衍生等问题。本文通过对提取、净化、测定及确证等条件的研究及优化,建立了检测酶制剂中3-NPA的固相萃取/超高效液相色谱电喷雾串联质谱方法,可实现对3-NPA的定性及定量分析。
1 实验部分
1.1 仪器、试剂与材料
Waters ACQULTY UPLC-Quattro Premier XE液相色谱-质谱联用仪(美国Waters公司);冷冻离心机(美国Beckman公司);漩涡混合仪(德国IKA公司);LABOROTA4001旋转浓缩仪(德国Heidolph公司);Milli-Q超纯水仪(美国Millipore公司)。
3-NPA(97.5%,Dr.Ehrenstorfer公司);乙腈(色谱纯,Fisher公司);氯化钠、无水硫酸钠均为分析纯,无水硫酸钠使用前于600℃马弗炉中烘烤5 h后密封保存。PSA固相萃取柱(3 mL,500 mg,Supelco公司)。
3-NPA标准储备液的配制:称取标准品10.0 mg于10 mL棕色容量瓶,用甲醇溶解并定容至10 mL,配成1.0 g/L的标准储备液,于-20℃下保存。3-NPA标准工作溶液的配制:取上述储备液0.025 mL于棕色容量瓶中,用甲醇定容至25 mL,配制成1.0 mg/L的标准工作溶液。
1.2 样品前处理
1.2.1 液体酶制剂中3-NPA的提取 称取5.00 g酶制剂样品于50 mL离心管中,加入20 mL乙腈振荡20 min后,再加入5 g氯化钠混合均匀后振荡10 min,以4 500 r/min离心5 min。取上清液10 mL,待净化。
1.2.2 固体酶制剂中3-NPA的提取 称取5.00 g酶制剂样品于50 mL离心管中,加入20 mL乙腈振荡20 min后,以4 500 r/min离心5 min。取上清液10 mL,待净化。
1.2.3 净 化 PSA固相萃取柱上加1.0 g无水硫酸钠后用6 mL乙腈活化,将试样溶液过柱,用10 mL甲醇洗脱,收集洗脱液。经氮气吹至近干后,用0.5% 甲酸水溶液定容至1.0 mL,过0.22 μm滤膜后,待测。
1.3 仪器条件
色谱条件:色谱柱Waters HSS T3柱(1.8 μm,2.1 mm×100 mm)。流动相为乙腈(A)和水(B),梯度洗脱程序:0~0.5 min,3% ~10%A;0.5~2.0 min,10% ~80%A;2.0~2.8 min,80%A;2.8~3.5 min,80% ~3%A;3.5~4.5 min,3%A。流速0.3 mL/min,进样体积5.0 μL,柱温40℃。
MS/MS条件:离子源为电喷雾电离源,负离子模式(ESI-),毛细管电压为2.91 kV;离子源温度115℃;脱溶剂气温度:450℃;脱溶剂气流量500 L/h;碰撞气为氩气,流量为0.2 mL/min;锥孔电压13 V;碰撞能量10 eV;检测模式为多反应监测(MRM);母离子为m/z117.9;特征离子为m/z46.3。
2 结果与讨论
2.1 前处理条件的优化
考察了甲醇、乙腈、乙酸乙酯等不同有机溶剂的提取效果。结果表明,甲醇的提取率为90%但干扰较大,乙酸乙酯的提取率仅为70%,而乙腈的提取率达到95%且干扰较少。同时在提取过程中加入氯化钠将水相和有机相分离。适量的氯化钠在离心后加入有助于减少有机溶剂中的漂浮物,使样品更清澈,有利于固相萃取的进行。
2.2 固相萃取柱的选择
据文献报道[12],可采用乙酸乙酯和三氯甲烷液液萃取的方式提取净化甘蔗中的3-NPA。但该方法的溶剂使用量大,且多次萃取容易造成待测物损失。本研究中采用固相萃取技术完成目标化合物的富集及净化。实验考察了不同萃取柱(Oasis WAX、PSA及氨基柱)的净化效果,结果显示,对酸性物质有较好选择性的Oasis MAX柱[13]虽能将目标物完全保留在小柱上,但无合适的洗脱液将目标物洗脱下来。
实验中比较了特征相似的氨基柱和PSA柱的净化效果。使用加标的乙腈溶液上固相萃取柱后,收集上柱溶液进行分析,3-NPA完全保留在氨基柱和PSA柱上。研究表明当强极性化合物在氨基柱上吸附力太强时可考虑用PSA柱代替[14]。比较了3-NPA的乙腈溶液在氨基柱和PSA柱上的解吸能力,在实验中用不同的洗脱液分别洗脱吸附在两种固相萃取柱上的3-NPA(见图1)。实验结果表明,过PSA固相萃取柱的回收率高于氨基柱,可能是由于目标化合物3-NPA在氨基柱上吸附过强。因此实验选择PSA柱为固相萃取柱进行进化。
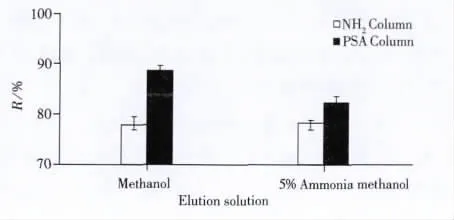
图1 3-NPA在不同固相萃取柱上的解离图Fig.1 Figure of dissociation of 3-NPA in different solid phase extraction columns
2.3 PSA固相萃取条件的优化
为了提高净化效果和降低基质效应,一般会对固相萃取柱进行淋洗。实验考察了不同体积比(10∶90、12∶88、20∶80)的异丙醇-乙腈作为淋洗液时3-NPA的损失情况。结果表明,用上述淋洗液后3-NPA均有损失。因此实验不采用淋洗步骤,而通过基质标准曲线降低基质效应的影响。
吸附于PSA固相萃取柱上的目标化合物,需要强极性溶剂进行洗脱。实验考察了乙腈、甲醇以及不同体积比的乙腈-甲醇(50∶50、30∶70、10∶90)溶液的洗脱能力,实验结果表明,甲醇比例越高,吸附在PSA柱上的3-NPA越容易洗脱下来。最终选择100%甲醇作为洗脱溶液。进一步考察了甲醇用量(3、5、8、10、15、20 mL)的影响,结果显示当洗脱溶液体积达到10 mL时,3-NPA已被全部洗脱下来,最终确定10 mL甲醇为洗脱溶液。
2.4 色谱柱的选择
文献报道使用反相色谱柱分离3-NPA时,由于3-NPA具有很强的酸性和亲水性,在反相柱上保留较弱,因此需要使用磷酸二氢钾溶液作为流动相[15]。由于液质联用不建议用磷酸盐等不挥发性物质为流动相,因此实验重点考察了有利于极性物质保留的反相色谱柱Waters ACQUITY HSS T3、Waters ACQUITY C18、Aglient Eclipse Plus C18、Waters UPLC BEH Amide及亲水性色谱柱Waters ACQUITY BEH HILIC对3-NPA的分离效果(见图2)。结果表明,3-NPA在Waters ACQUITY C18及Aglient Eclipse Plus C18的保留时间均在0.7 min,且峰形拖尾,在Waters ACQUITY BEH HILIC及Waters UPLC BEH Amide上的峰形尽管有所改善但仍有拖尾。而3-NPA在Waters ACQUITY HSS T3上的保留时间在1.5 min,且峰形良好。因此选择Waters ACQUITY HSS T3作为3-NPA的分析柱。

图2 3-NPA在不同色谱柱上的保留色谱图Fig.2 Chromatograms of retention of 3-NPA on different columns A.Waters ACQUITY C18(1.8 μm,2.1 mm ×100 mm);B.Aglient Eclipse Plus C18(1.8 μm,2.1 mm ×100 mm);C.ACQUITY BEH HILIC(1.8 μm,2.1 mm ×100 mm);D.Waters UPLC BEH Amide(1.7 μm,2.1 mm ×100 mm);E.Waters ACQUITY HSS T3(1.8 μm,2.1 mm ×100 mm)
2.5 流动相条件的优化
由于3-NPA是一种小分子有机酸,具有较强的酸性和亲水性。因此主要考察了甲醇-水、乙腈-水、乙腈-5 mmol/L乙酸铵溶液体系的分离效果。结果表明,乙腈-水为流动相时的响应最高,且保留时间在1.5 min以后。流动相中加盐后灵敏度降低,可能是盐的存在抑制了目标化合物的电离。因此确定乙腈-水为最佳流动相。
为了增强目标化合物在色谱柱上的保留。实验比较了纯水、0.1%氨水溶液、0.05%甲酸水溶液、0.5%甲酸水溶液、5 mmol/L甲酸铵溶液作为定容液时对目标化合物响应的影响。实验发现,用0.5%甲酸水溶液作为定容溶液时不仅保留时间明显增强,且响应也明显增强。最终确定0.5%甲酸水溶液作为定容溶液。
2.6 基质干扰的影响
由于酶制剂大多由真菌菌种发酵而成,含有大量蛋白,因此对3-NPA的提取及测定有干扰,且无法得到3-NPA的同位素内标。因此选择基质加标工作曲线进行外标法定量,对基质效应及净化过程中的损失进行校正。以果胶酶为例,不采用果胶酶基质标准时,加标5.0 μg/kg的回收率为32.1%~42.1%,而采用基质标准后,回收率为63.2%~79.6%。回收率提高了1倍。实验还比较了不同酶制剂为基质标准对3-NPA检测的干扰校正情况(见图3),不同酶制剂对3-NPA的干扰影响不同,果胶酶对3-NPA的抑制干扰最大,果胶超滤酶的干扰较小,因此实验需要对不同品种的酶制剂进行基质标准校正,进而进行定量分析。

图3 不同酶制剂的基质影响效应Fig.3 The matrix effects of different zymins
2.7 方法验证参数
对不同种类的酶制剂绘制基质标准曲线。分别称取6份5.0 g空白酶制剂基质于50 mL离心管中,分别加入1.0 μg/L标准工作溶液25、50、100、150、250 μL,进行前处理后在优化条件下测定。以3-NPA的峰面积(Y)对相应的质量浓度(X,μg/L)绘制基质标准工作曲线。得出回归方程、相关系数、线性范围及定量下限(S/N≥10)(见表1)。结果显示,3-硝基丙酸在6种酶制剂基质中的线性范围均为12.5 ~125 μg·L-1,相关系数大于 0.993,定量下限为 5.0 μg/kg。
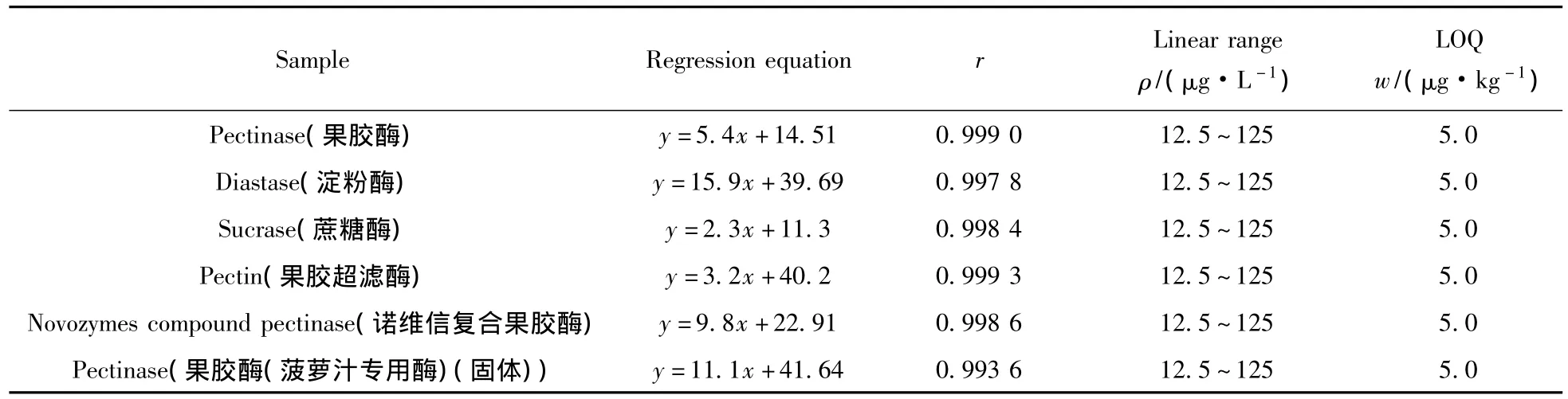
表1 不同酶制剂的回归方程、相关系数(r)、线性范围及定量下限Table 1 Regression equation,correlation coefficient(r),linearity range and detection limit in different zymin
分别称取不含3-NPA的空白果胶酶、果胶超滤酶、诺维信复合果胶酶、蛋白酶、蔗糖酶样品进行方法回收率和精密度试验。加入的3-NPA标准品浓度分别为5.0、10.0、20.0 μg/kg,每种浓度做5次平行,按优化方法进行实验。每种样品的平均回收率及相对标准偏差(RSD)见表2。在3个加标水平下,3-硝基丙酸的回收率为69.1%~114.4%,RSD为6.9%~8.9%。
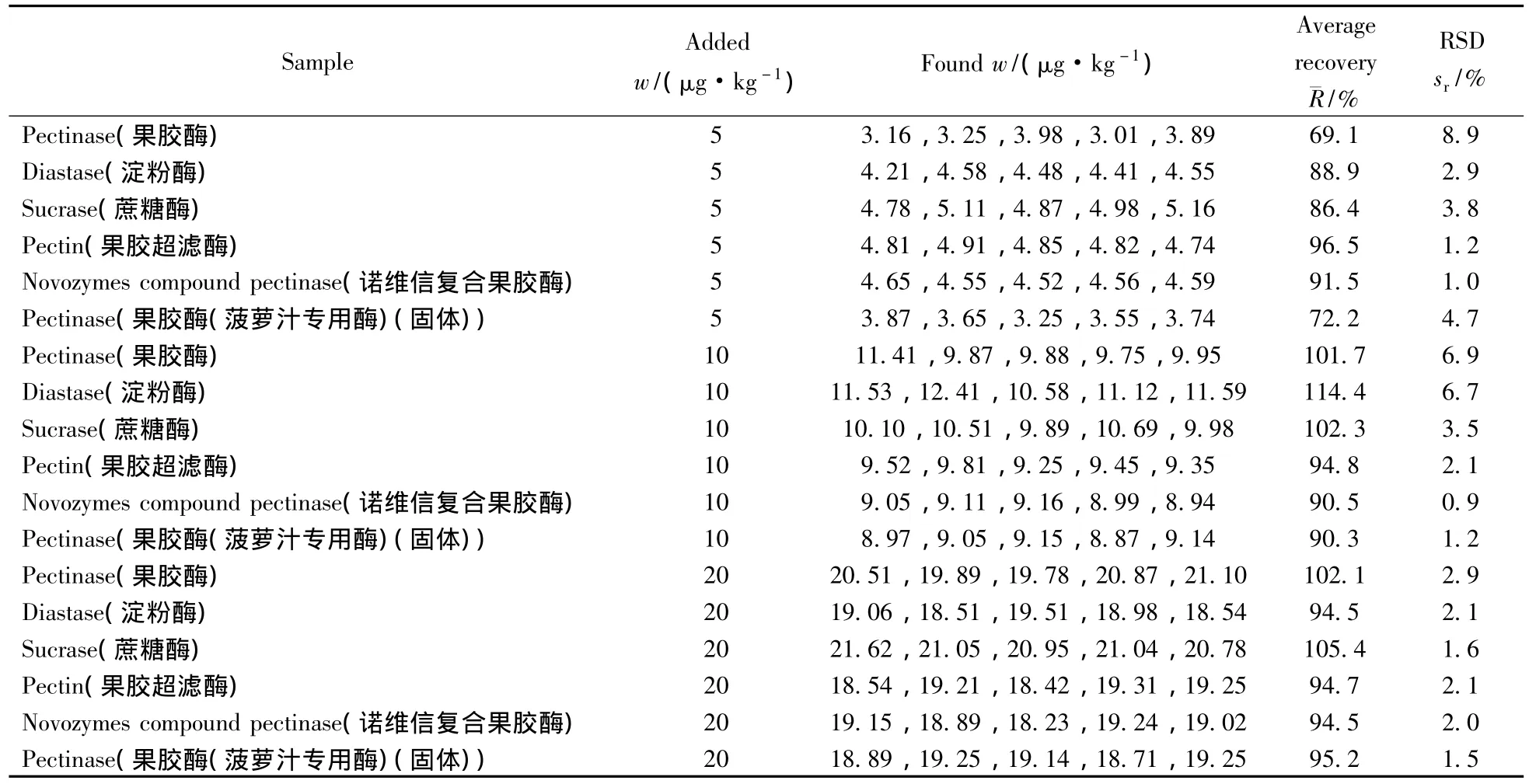
表2 3-NPA在不同种类酶制剂的加标回收率及相对标准偏差Table 2 Recovery and precision of 3-NPA in different enzymic preparations
2.8 方法应用
为了考察本方法的有效性与实用性,采购7种常用的酶制剂样品进行3-NPA的检测。结果表明,所有样品中均未检出3-NPA残留。图4为酶制剂实际样品的色谱图。
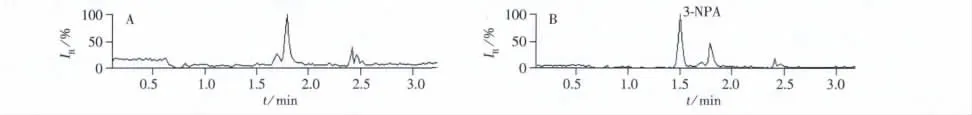
图4 果胶酶样品(A)及样品加标(B)的色谱图Fig.4 Chromatograms of pectinase sample(A)and sample spiked with 3-NPA(B)
3 结论
本文建立了酶制剂中3-NPA的超高效液相色谱-电喷雾串联质谱检测方法。样品经乙腈提取,固相萃取柱净化,质谱确认后,可实现对3-NPA的定性及定量分析。该方法前处理过程简便、灵敏度高、选择性好,可满足日常检测的需要,为酶制剂的质量安全提供了技术保障。
[1] Leza H A R,Jasso R M R,Esquivel J C C,Herrera R R,Aguilar C N.Biohem.Eng.J,2012,65:90-95.
[2] Carrín M E,Ceci L N,Lozano J E.Food Chem.,2004,84:173 -178.
[3] Tribst A A L,Cristianini M.Innovative Food Science and Emerging Technologies,2012,13:107 -111.
[4] Li F,Xu Q Y.Lab.Animal Sci.(李峰,徐群渊.实验动物科学),2009,26(4):12-15.
[5] Liu H G,Ma Y,Yang A C,Meng D W,Zhang Y,Zhang J G.Chinese Journal of Minimally Invasive Neurosurgery(刘焕光,马羽,杨岸超,孟大伟,张颖,张建国.中国微侵袭神经外科杂志),2012,17(7):319-321.
[6] Olsen C,Rustad A,Fonnum F,Paulsen R E,Hassel B.Brain Res.,1999,850:144 -149.
[7] Luchowski P,Luchowska E,Turskia W A,Urbanskaa E M.Neurosci.Lett.,2002,330:49 -52.
[8] Liu Y,Wang Y H,Liu X J,Li X F,Liu Z H,Hu W J.J.Hyg.Res.(刘勇,王玉华,刘兴玠,李秀芳,刘增辉,胡文娟.卫生研究),1989,18(5):38-40.
[9] Shao G J,Han J K,Wu D Q.Chin.J.Health Lab.Technol.(邵国健,韩建康,吴丹青.中国卫生检验杂志),2012,22(4):711-712.
[10] Jiang T,Zhang Q L,Luo X Y.J.Hyg.Res.(江涛,张庆林,罗雪云.卫生研究),1999,28(5):300-302.
[11] Wang J,Lei Z Y,Feng X Q.Pratacultural Science(汪儆,雷祖玉,冯学勤.草业科学),1992,9(2):34-37.
[12] WS/T 10 -1996.Diagnotic Criteria and Principles of Management for Food Poisoning of Mildew Sugareane.Standards of Ministry of Health of the Peoples Republic of China(变质甘蔗食物中毒诊断标准及处理原则.中华人民共和国卫生部标准).
[13] Li B,Wu G H,Liu W,Zhao X D,Zhao H Y,Xue Y,Zhao R.Chin.J.Food Hyg.(李兵,吴国华,刘伟,赵旭东,赵海燕,薜颖,赵榕.中国食品卫生杂志),2012,24(2):127-132.
[14] Chen X H,Wang Q J.Technology and Application of Solid-phase Extraction.Beijing:Science Press(陈小华,汪群杰.固相萃取技术与应用.北京:科学出版社),2010:56.
[15] Gong X M,Ren Y P,Dong J,Sun J,Li J,Jin C,Yu J L.J.Instrum.Anal.(宫小明,任一平,董静,孙军,李健,金超,于金玲.分析测试学报),2011,30(1):6-12.
猜你喜欢
中国油脂(2020年3期)2020-04-10
中成药(2018年7期)2018-08-04
猪业科学(2018年5期)2018-07-17
无机化学学报(2016年8期)2016-12-06
广东饲料(2016年2期)2016-12-01
化学分析计量(2016年1期)2016-03-14
分析测试学报(2015年3期)2016-01-13
化工进展(2015年6期)2015-11-13
中国果菜(2015年2期)2015-03-11
中国酿造(2014年9期)2014-03-11
