
双碳源在LiFePO4/C微波固相合成中的协同效应
2013-11-20 01:23闫出博杨建瑞解勤兴张宗杰尹翠玉张宇峰
河南工程学院学报(自然科学版) 2013年3期
闫出博,杨建瑞,解勤兴*,张宗杰,解 超,尹翠玉,张宇峰
(1.天津工业大学 材料科学与工程学院, 天津 300387;2.山东理工大学 材料科学与工程学院,山东 淄博 255049)
磷酸铁锂正极材料理论比容量可达170 mAh/g,呈现较高的工作电压(3.4 V,vs.Li)和稳定的充放电电压平台,同时具有良好的循环性能、原材料来源丰富、成本低、安全无毒等特点[1].自1997年Goodenough等发现橄榄石型LiFePO4可作为锂离子电池正极材料以来,LiFePO4已经成为新能源研究领域的热点[2-3].
目前合成LiFePO4的方法有多种,主要包括高温固相法[4]、水热合成法[5]、溶胶凝胶法[6]、液相共沉淀法[7]和碳热还原法[8]等.目前,高温固相法因工艺简单在工业化生产中占据重要地位,但这种方法生产周期长、能耗大,从而导致磷酸铁锂的生产成本居高不下.微波法是近年来新兴的合成技术,具有加热时间短、物料受热均匀、节能高效等优点,因而磷酸铁锂的微波合成得到了越来越多的关注[9-10].同时,目前大多数合成研究直接采用或改装家用微波炉进行,不易控制反应温度,工艺参数的量化结果无法直接应用于工业化生产.本研究分别以葡萄糖与葡萄糖/乙炔黑为碳源,以高温固相法中广泛使用的Li2CO3,FeC2O4·2H2O和NH4H2PO4为前驱体,利用湖南中昇热能科技有限公司生产的RWS型微波真空高温实验炉制备LiFePO4/C正极材料,实验过程中利用美国雷泰公司产红外线测温仪进行精确控温,系统研究了该微波合成体系中乙炔黑及反应温度对LiFePO4/C微结构和电化学性能的影响.
1 实验部分
1.1 实验材料及仪器
本实验所用Li2CO3,FeC2O4·2H2O,NH4H2PO4和葡萄糖均为分析纯,反应在RWS型微波真空高温实验炉中进行.
1.2 LiFePO4/C的合成
Li2CO3,FeC2O4·2H2O和NH4H2PO4以1∶2∶2的摩尔比(满足LiFePO4分子中Li,Fe,P的原子数比例=1∶1∶1)混合均匀,并加入10%的葡萄糖作为碳源.将混合物料以丙酮作介质在氩气氛中以200 r/min球磨4 h.干燥后压制成0.5 cm厚的圆片,置于微波炉内的刚玉坩埚中.在氮气氛下,快速升温至目标温度(分别为600 ℃,650 ℃,700 ℃)并持续反应40 min.将反应后得到的产物粉碎得到黑色的LiFePO4/C正极材料,分别标志为600-LFP/C1,650-LFP/C1与700-LFP/C1.
Li2CO3,FeC2O4·2H2O和NH4H2PO4以1∶2∶2的摩尔比混合均匀,并加入10%的葡萄糖和1%的乙炔黑(电池级).乙炔黑在作为碳源的同时又可以作为微波吸收增强剂,反应过程同上.将得到的黑色LiFePO4/C正极材料分别标志为600-LFP/C2,650-LFP/C2和700-LFP/C2.
1.3 材料表征及电化学性能测试
按照文献[11]报道的方法测定磷酸铁锂正极材料的含碳量,用日本Rigaku RINT2000 X射线粉末衍射仪测定材料的X射线粉末衍射图谱(Cu靶,Kα,λ=0.154 06 nm,电流=150 mA, 电压=40 kV,扫描范围为10°~80°(2θ), 步长为0.02°),用日本Hitachi S-4800扫描电子显微镜(SEM)观察材料的微观形貌.材料的电导率用RTS-9型四探针测试仪(广州四探针电子科技有限公司)在25 ℃下进行测试,探针系数为6.28,平均间距为1 mm.测试前将材料研细后以 40 MPa压成直径为20 mm、厚度约4 mm的圆片.
电化学性质测定在扣式电池中进行.LiFePO4/C导电剂(乙炔黑)和黏结剂(PVDF)以80∶10∶10的质量比混合,用N-甲基吡咯烷酮(NMP)作溶剂研磨成均匀浆状.用AFA-Ⅲ型自动涂膜烘干机(美国MTI公司)将正极浆料均匀地涂布于铝箔之上,110 ℃干燥后切成直径为13 mm的圆形正极片.以金属锂片为负极,CeIgard 2300为隔膜,1 M LiPF6/EC+DMC(体积比为1∶1)为电解液在氩气氛手套箱中组装2430型扣式电池.采用CT2001A型电池测试系统(武汉蓝电)进行充放电测试,测试温度为25 ℃,电压上限为4.2 V,下限为2.0 V.
2 结果与讨论
2.1 材料表征
图1为不同条件下合成的LiFePO4/C的X射线衍射谱图(XRD).从XRD谱图可以看到它们均为单一物相的橄榄石型磷酸铁锂,不含杂质的衍射峰而且材料具有很好的结晶度.LiFePO4的平均晶粒大小用Debye-Scherrer公式(D= 0.9λ/βcosθ,其中D为晶粒直径,λ为X射线波长,θ为Bragg衍射角,β为衍射峰的半峰宽)计算得出,结果列于表1.同时,通过精修得到的晶胞参数和晶胞体积列于表2.由表1可见仅以葡萄糖作碳源(以下称单碳源)合成的LiFePO4晶粒大小在75~77 nm;以葡萄糖/乙炔黑作碳源(以下称双碳源)合成的LiFePO4晶粒粒径较大,在80~87 nm,原因可归结为乙炔黑较强的微波吸收能力,使起始物料的微波加热效果较前者好,在相同的反应条件下,双碳源合成的LiFePO4具有更好的结晶度.由表2可见,LiFePO4的晶胞参数和晶胞体积与温度有关,600 ℃合成的较大,650 ℃和700 ℃合成的较小而且数值接近,这可能是在较高的温度下锂盐的挥发使LiFePO4晶格中形成锂位的缺陷造成的.
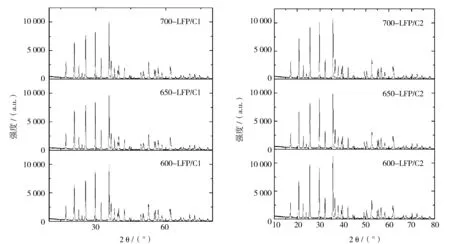
图1 LiFePO4/C正极材料的粉末X射线衍射(XRD)图谱Fig.1 X-ray powder diffraction patterns (XRD) of LiFePO4/C cathode materials

材料电导率/(S·cm-1)晶粒粒径/nm碳含量/%600-LFP/C13.48×10-477.44.3650-LFP/C15.41×10-475.04.7700-LFP/C13.20×10-477.04.1600-LFP/C27.42×10-486.57.3650-LFP/C211.90×10-482.18.5700-LFP/C215.50×10-479.95.8
由表1中复合材料的碳含量测试结果可知,单碳源LiFePO4/C产物的碳含量非常接近,在4.1%~4.7%,而双碳源LiFePO4/C产物因原材料中的乙炔黑致使碳含量要高一些,差别也比较大(在5.82%~8.47%).同时,LiFePO4/C的碳含量也与合成温度有关,650 ℃合成的LiFePO4/C因反应温度适中而使碳化反应比较完全,其碳含量在同一体系3种材料当中为最高;600 ℃合成的LiFePO4/C碳含量较650 ℃的为低,可能因为该反应温度不足以使全部葡萄糖在较短时间内完全分解碳化,在碳含量的测试过程中未分解部分溶解于水而导致结果偏低;700 ℃合成的LiFePO4/C碳含量最低,可能是其中小部分碳在较高温度下与体系中的微量氧发生反应生成CO和/或CO2气体而造成碳的损失.通过对比LiFePO4/C的电导率可以发现,材料的电导率与所含的碳有关.LFP/C2体系因为含乙炔黑,因而导电性要LFP/C1体系的好.同时可以看到除700-LFP/C2之外,材料的电导率随着含碳量的增加而提高.700-LFP/C2比同体系中的其他两种材料的含碳量低,但是其电导率达15.50×10-4S/cm为最高.除误差因素外,还可能有极少量非晶态的碳(乙炔黑和葡萄糖分解产生的碳)在较高的微波温度下发生一定程度的微晶化,随着温度的升高微晶的生长趋于更加有序化,甚至可生成接近石墨形态的微晶.微晶晶体中相邻原子的内外层轨道靠近产生不同程度的重叠,致使电子在原子间的迁移能垒降低,从宏观上看整个材料体系的电导率增大.关于碳材料碳化温度与导电性的关系已有不少报道[12-13].但是微晶碳的含量低,不足以在XRD图谱上发现衍射峰.
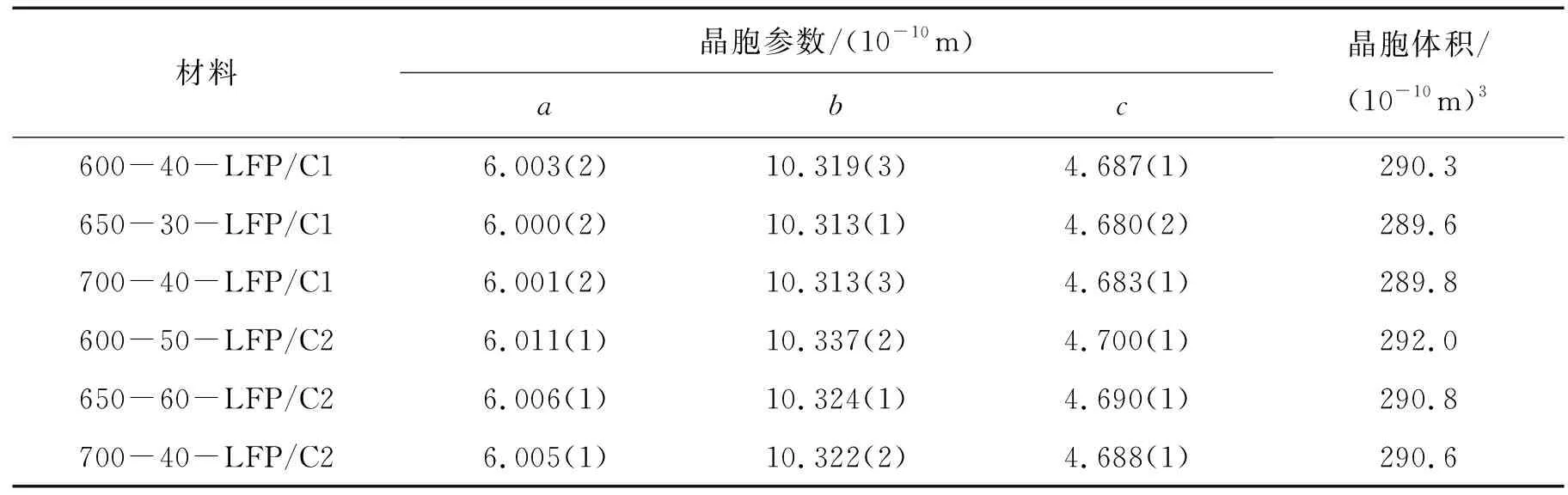
表2 LiFePO4的晶胞参数及晶胞体积Tab.2 The refined lattice constants and cell volume of LiFePO4
所合成的LiFePO4/C材料的扫描电镜(SEM)图见图2.显然,粉体的直径均明显大于由Scherrer公式计算得到的晶粒粒径,说明这些大的颗粒是由小的晶粒聚集而成.同时,单碳源和双碳源LiFePO4/C的微观形貌也有所不同:
(1)单碳源600-LFP/C1颗粒大小不一,部分颗粒表面有纤维或膜状物质,可能是葡萄糖分解后形成的碳纤维和碳膜包覆在磷酸铁锂的表面.将粒径大的颗粒放大观察可以发现是它是由较小的颗粒聚集而成.650-LFP/C1中有少量直径为5~6 μm的球形颗粒,放大观察发现有更多的的纳米球形颗粒(200~700 nm大小不等)分布在大的颗粒表面,这是因为起始物料的微波吸收能力较差,致使产物局部温度过高而熔融成小的微球.700-LFP/C1颗粒形状不规则,没有类似650-LFP/C1中的微球,粒径分布较为均匀,表示反应比较彻底,物相比较均一.
(2)由于乙炔黑的协同作用,双碳源LiFePO4/C产物呈现不同的微观形貌.乙炔黑的加入在一定程度上提高了物料的微波吸收能力,使物料可以均匀受热.600-LFP/C2的颗粒形状不规则,将较大的颗粒放大观察可以发现其中团聚的小颗粒具有明显的晶界.650-LFP/C2因温度升高粒径明显增大,颗粒形状接近于球形,表面光滑,烧结现象明显.700-LFP/C2因温度进一步升高而产生熔融现象,球形颗粒周围有类似凝胶状的物质,应该是熔融的磷酸铁锂冷却固化后的形态.

图2 扫描电镜(SEM)Fig.2 Scanning electron microscopy (SEM)
2.2 LiFePO4/C的电化学性能
LiFePO4/C正极材料的0.1C(1C为LiFePO4的理论比容量170 mAh/g)首次充放电曲线见图3.由图3可见材料均具有稳定的充放电平台,分别约为3.5 V和 3.4 V,电压差小于0.1 V.不同碳源、不同温度合成的LiFePO4/C呈现不同的电化学性能.仅用葡萄糖作碳源合成的600-LFP/C1,650-LFP/C1和700-LFP/C1的首次充电比容量分别为126.76 mAh/g,107.51 mAh/g和131.05 mAh/g,放电比容量分别为94.30 mAh/g,92.32 mAh/g和110.13 mAh/g,显然700-LFP/C1的0.1C充放电性能最好.在反应物中添加乙炔黑后产物的0.1C比容量除700-LFP/C2外均有大幅度提升,其首次充电比容量分别达到149.8 mAh/g(600 ℃),134.3 mAh/g(650 ℃)和125.4 mAh/g(700 ℃),放电比容量分别为131.9 mAh/g,116.2 mAh/g和109.4 mAh/g,其中600-LFP/C2的0.1C充放电性能最好,700-LFP/C2因为存在过烧现象导致比容量有所下降,这与扫描电镜分析结果一致.
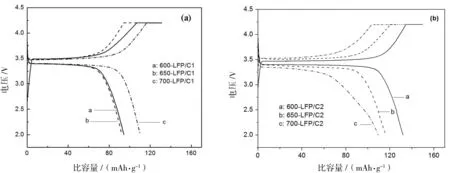
图3 (a) LFP/C1 和 (b) LFP/C2的0.1C首次充放电曲线Fig.3 The initial charge/discharge (c/d) curves of (a) LFP/C1 and (b) LFP/C2
两种体系的LiFePO4/C材料0.2C充放电循环性能曲线比较见图4.显然,在起始阶段放电比容量随着循环次数的增加而增大,在循环10~20次后电极材料得到充分活化后达到最大值.单碳源制备的LiFePO4/C中,700-LFP/C1的电化学性能较好,其0.2C最大放电比容量为125.2 mAh/g,循环100次后达到 120.9 mAh/g,保持率为96.6%.而600-LFP/C1和650-LFP/C1的0.2C放电容量相对较低而且相似,经100次循环后与最高值(分别为107.5 mAh/g和104.0 mAh/g)比较仅略有衰减.双碳源LiFePO4/C的0.2C放电容量也明显增大,电化学活性最高的为600-LFP/C2,其次为650-LFP/C2和700-LFP/C2.三种LFP/C2材料经100次循环后放电比容量分别达到139.6 mAh/g,126.1 mAh/g和118.2 mAh/g,最大值分别为140.6 mAh/g,127.7 mAh/g和121.3 mAh/g.可以看出添加乙炔黑后合成的LiFePO4/C的充放电循环性能得到很大提高,100次循环后放电比容量几乎没有衰减.
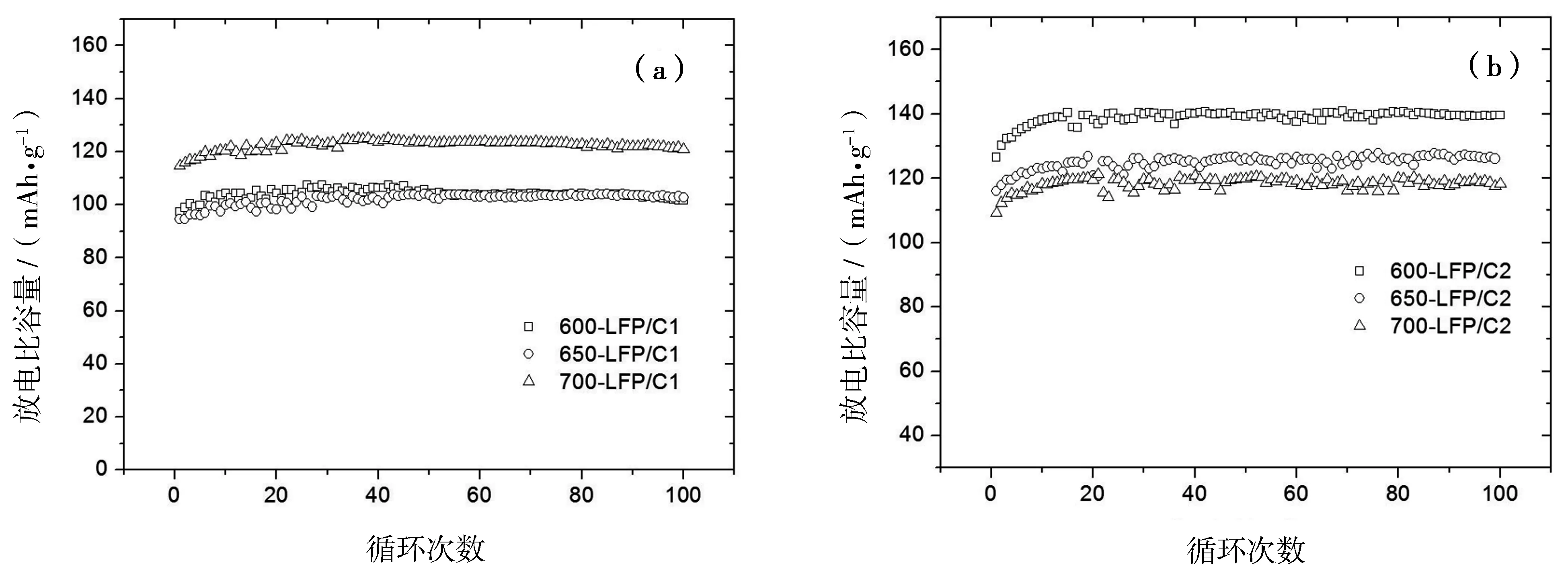
图4 (a) LFP/C1和(b) LFP/C2的0.2C充放电循环性能曲线Fig.4 Cycling performance of (a)LFP/C1 and (b)LFP/C2 at a c/d rate of 0.2C
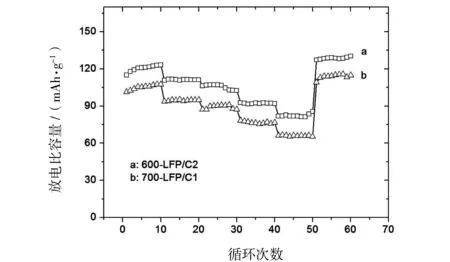
图5 LiFePO4/C的倍率放电性能(充电均为0.2C,放电为0.2C~8C)Fig.5 Discharge capacity retentions of LiFePO4/C at various discharge rates from 0.2C to 8C (the charge was all performed at 0.2C)
700-LFP/C1和600-LFP/C2两种性能较好的复合材料在不同倍率下的放电性能曲线见图5.每次循环均以0.2C进行充电,然后分别以0.2C,1C,2C,4C,8C,0.2C 的倍率依次进行连续阶段放电(不同倍率各循环10次),截止电压为2.0~4.2 V.由图5可见,倍率越大比容量越小,但是经过大倍率充放电循环之后,再以0.2C充放电,材料的比容量能够得到恢复.
(1)单碳源LiFePO4/C以0.2C充放电10次后放电比容量达到110.8 mAh/g.经过大倍率放电,再次以0.2C进行充放电,其比容量可达到115.7 mAh/g.显然,材料经过大倍率放电后容量不仅没有衰减,反而有小幅度上升,呈现良好的大倍率循环性能,其8C首次放电比容量(78.3 mAh/g)可达到0.2C首次放电容量的77%左右.
(2)双碳源LiFePO4/C在0.2C充放电10次后放电比容量分别为123.2 mAh/g,经过大倍率阶段放电,再次以0.2C进行充放电后比容量达到130.3 mAh/g,其8C首次放电比容量(81.8 mAh/g)几乎接近0.2C首次放电容量的71%,双碳源产物的比容量明显优于单碳源产物.
3 结论
仅以葡萄糖作为碳源,由于起始物料的微波吸收能力弱而受热不均,在较低温度下合成的LiFePO4/C的微结构和电化学性能较差,700 ℃为较佳反应温度.而在碳源中加入乙炔黑后,600 ℃合成的LiFePO4/C即具有最佳电化学性能,温度过高反而易引起材料烧结团聚导致电化学性能降低.同时,双碳源LiFePO4/C的充放电性能要优于单碳源LiFePO4/C.显然,乙炔黑的加入不仅提高了目标材料的导电性,也有利于增强起始物料的微波吸收能力,易于使反应体系均匀受热,在较低温度下即可得到结构完善、物相均匀、电化学性能良好的LiFePO4/C正极材料.
参考文献:
[1] Zhang W J.Structure and performance of LiFePO4cathode materials:a review [J].Power Sources,2011,196(6):2962-2970.
[2] Padhi A K,Nanjundaswamy K S,Masquelier C,et al.Effect of structure on the Fe3+/Fe2+redox couple in iron phosphates [J].Electrochemistry Society,1997,144(5):1609-1613.
[3] Goodenough J B,Kim Y.Challenges for rechargeable Li batteries [J].Chemistry.Mater,2010,22 (3):587-603.
[4] Chung S Y,Bloking J T,Chiang Y T.Electronically conductive phospho-olivines as lithium storage electrodes [J].Nature Materials,2002,1(2):123-128.
[5] Chen J,Whittingham M S.Hydrothermal synthesis of lithium iron phosphate [J].Electrochemistry Communication,2006,8(5):855-858.
[6] Rho Y H,Nazar L F,Perry L,et al.Surface chemistry of LiFePO4studied by Mössbauer and X-ray photoelectron spectroscopy and its effect on electrochemical properties [J].Electrochemistry Society,2007,154(4):A283-A289.
[7] Jugovi C D,Mitri C M,Kuzmanovi C M,et al.Preparation of LiFePO4/C composites by co-precipitation in molten stearic acid [J].Power Sources,2011,196(10):4613-4618.
[8] Wang L,Liang G C,Ou X Q,et al.Effect of synthesis temperature on the properties of LiFePO4/C composites prepared by carbothermal reduction[J].Power Sources,2009,189(1):423-428.
[9] Beninati S,Damen L,Mastragostino M.MW-assisted synthesis of LiFePO4for high power applications [J].Power Sources,2008,180(2):875-879.
[10] Guo X F,Zhan H,Zhou Y H.Rapid synthesis of LiFePO4/C composite by microwave method [J].Solid State Ionics,2009,180(4/5):386-391.
[11] Zhao J,He J,Zhou J,et al.Facile synthesis for LiFePO4nanospheres in tridimensional porous carbon framework for lithium ion batteries [J].Physics Chemistry,2011,115(6):2888-2894.
[12] 江泽慧,张东升,费本华,等.炭化温度对竹炭微观结构及电性能的影响[J].新型炭材料,2004,19(4):249-253.
[13] Xu B,Wu F,Cao G P,et al.Effect of carbonization temperature on microstructure of PAN-based activated carbon fibers prepared by CO2activation [J].New Carbon Materials,2006,21(1):14-18.
猜你喜欢
环境工程技术学报(2022年3期)2022-06-05
昆钢科技(2021年6期)2021-03-09
安徽电子信息职业技术学院学报(2019年2期)2019-04-26
科普创作(2018年1期)2018-11-30
制造业自动化(2017年2期)2017-03-20
电源技术(2016年9期)2016-02-27
电源技术(2016年9期)2016-02-27
东北电力大学学报(2015年4期)2015-11-13
中国塑料(2015年6期)2015-11-13
自动化博览(2014年8期)2014-02-28
