
多羟基生物碱及其抗结核活性研究进展
2014-02-24 08:31刘春艳
科技视界 2014年11期
王 蕊 刘春艳
(河北联合大学 公共卫生学院,河北 唐山 063000)
1 结核病现状
作为一种传染病,结核病主要分为慢性与缓发两种,发病年龄多为15-35 岁,且治疗效果欠佳,预后差。 该病在上世纪又被称为痨病,并且有“十痨九死”的说法。 结核杆菌以呼吸道为媒介进行传播,病变部位主要在肺部,此外腹膜、脑膜、淋巴尤其是颈部淋巴也是其主要继发部位,结核病一般为慢性病程,以乏力、低热为主要全身症状,以咯血、咳嗽为呼吸系统的特异性症状,影像学主要表现为结核结节继发干酪样坏死和空洞。 作为人类健康的主要威胁因素之一,结核病曾在历史上大范围的广泛流行,世界卫生组织调查显示结核病新增患者每年约800 万,死亡人数更是高达200 万[1],远远超过了其他多种传染病如腹泻、疟疾及艾滋病的总死亡人数,随着现代医学进展尤其是临床诊断和制药水平的提高,自上世纪五十年代新一代抗结核药物如异烟肼(INH)、利福平(RFP)不断问世[2],为有效治疗结核病打下了坚实的基础。但从上世纪80 年代开始,不合理用药以及治疗的不彻底而产生的耐多药结核病(MDR-TB)和广泛耐药结核病(XDR-TB),以及结核杆菌与艾滋病毒等病原体共感染等问题[3],使得药物的抗结核效果显著降低,结核病发病率明显上升,因此深入研究结核病,探索新的靶向药物,既是一个医学相关性科学难题,又是一个关系到全人类身体健康的重大社会问题,绝对不容忽视。
2 多羟基生物碱
多羟基生物碱(Polyhydroxylated alkaloids),作为一种天然非化合产物,其在动物、植物及微生物体中广泛存在,是一种重要的糖苷酶抑制剂。多羟基生物碱可以转化为糖苷酶配体的天然类似物并通过与相应活性位点特异性结合,发挥抑制效应,多羟基生物碱有三个别称,分别为亚氨基环多醇(Iminocyclitol)、氮杂糖(Azasugars)、糖类生物碱(SugarShapedA1ka1oids)。 其在结构上与单糖类似,单糖环中的氧原子被氮原子替换后就可以形成多羟基生物碱,由于含氮杂环的种类繁多,羟基的数目以及立体化学也不同, 使得多羟基生物碱有着巨大的分子多样性。 迄今为止,已经有超过100 个的多羟基生物碱被分离得到。
依据含氮杂环具体结构,多羟基生物碱共分为多羟基哌啶,多羟基吡咯烷,多羟基双稠吡咯烷,多羟基去甲莨菪烷及多羟基吲哚里西啶生物碱五大类。
2.1 多羟基哌啶(Piperidine)生物碱
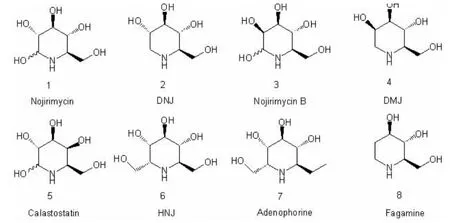
图1 多羟基哌啶生物碱
Inouye 等(日本)最早于1996 年就成功获得Nojirimycin 1,该物质来源于链霉菌,是一种链霉菌相关抗生素。 它是第一个纯天然多羟基哌啶类生物碱,能够介导葡萄糖苷酶活性的降低及最终失活[4]。 随后Nojirimycin B 3(甘露糖型野尻霉素N)[5]及Galactostatin 5(半乳糖型野尻霉素)[6]也相继从链霉菌中获得,但相关C-1 位被羟基取代的化合物不是很稳定,不便于分离和储存。 但通过催化氢化或者硼氢化钠等还原条件,可以把它们转化成为1-脱氧衍生物,来提高其稳定性。
在发现微生物中含有多羟基生物碱后不久,1974 年,人们从寥科植物荞麦Fagopyrum esculentum 中分离出首个植物来源的多羟基哌啶生物碱Fagamine 8[7]。 随后, 人们从桑树的树根中得到DNJ 2[8]-Moranoline。该物质被以后的研究证实也广泛存在于细菌如链霉菌、杆菌中。 DNJ 可以充分抑制包括蔗糖酶、海藻糖酶、转化酵素、麦芽糖酶及异麦芽糖酶在内的多种α-葡萄糖苷酶的活性, 而对葡萄糖苷酶I和II,β-葡萄糖苷酶,α-甘露糖苷酶,α-岩藻糖苷酶,α-半乳糖苷酶和β-半乳糖苷酶抑制活性较弱;DNJ 的C-2 差向异构体DMJ 4 (中文名:甘露糖型-1-脱氧野尻霉素)首先从豆科植物Lonchocarpus 中分离到,之后,人们发现它与DNJ 的C-1 位羟甲基取代物HNJ 6(中文名:高野尻霉素)共存于大戟属植物Omphalea diandra 中[9]。 2005 年Crews从马达加斯加岛的海绵体获得了Batzellasides 这一多羟基哌啶生物碱,该生物碱主要通过较长的烷基链取C-1 进行合成,这也是首次从海洋生物中分离到多羟基生物碱[10]。
2.2 多羟基吡咯烷(Pyrrolidine)生物碱
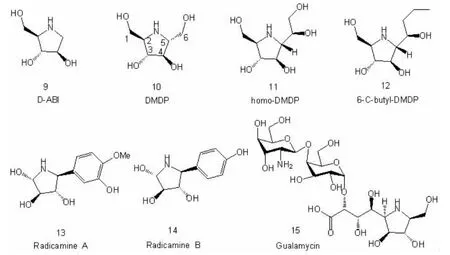
图2 多羟基吡咯烷生物碱
通过对毛鱼藤树叶的分离,人们于1976 年发现了DMDP 10(全称:2,5-Dihydroxyrnethyl-3,4-dihydroxypyrrolidine)[11], 随后又从不同种类的植物或微生物中提取得到[12]。 DMDP 在结构上与β-D-呋喃果糖相似,它对β-葡萄糖苷酶,α-葡萄糖苷酶,人β-木糖苷酶、β-半乳糖苷酶、溶酶体β-甘露糖苷酶产生强效抑制。从被发现之日起,DMDP就作为多羟基生物碱领域的模型化合物,吸引了化学家和生物化学家们的广泛关注和深入研究。 脱羟甲基DMDP 衍生物1,4-Dideoxy-1,4 -imino -D -arabinitol (D -ABI 9) 首 先 发 现 于 Angylocalyx boutiqueanus 的果实中[13],后来也从别的植物中分离得到,对α-葡萄糖苷酶有较强抑制性, 它同DMDP 一样也是热带和温带植物普遍存在的二级代谢物。
Nash 等通过分离野风信子Hyacinthoides non-scripta, 于 1997 年率先从其叶子中分离到 2,5-Dideoxy-2,5-imino-D-glycero-D-manno-heptitol (homoDMDP 11)[14], 随后人们又从风信子 Hyacinthus orientali 的球茎和绵枣儿Scilla campanulata 中分离得到。 HomoDMDP对多种糖苷酶(海藻糖苷酶、β-半乳糖苷酶、β-葡萄糖苷酶)均强效抑制。最新研究证实,6-C-butyl-DMDP 12 对淀粉葡萄糖苷酶及β-葡萄糖苷酶抑制性强,该物质主要由Adenophora triphylla var.japonica 风铃分离得到,丁基取代DMDP 6 号碳原子最终合成,遗憾的是,到目前为止其C-6 位的相对构型仍未确定[15]。
2001 年,Kusano 等人[16]从半边莲植物 Lobelia chinensis Lour 中分离到含芳香环的多羟基吡咯烷Radicamine A 13 和Radicamine B 14,并发现它们对α-葡萄糖苷酶有很好的抑制性。 有趣的是,与其它全R型的多羟基吡咯烷不同,它们的绝对构型被定为全S 型。 而经过某些科学家研究证明,它的绝对构型和其它的多羟基吡咯烷一样,均为全R 型[17]。
其实,上述多羟基生物碱其羟基被不同程度糖基化的产物也经常被分离到,例如通过对链霉素(Streptormyces sp.NK11687)分离,科研人员于1995 年成功获得Gualamycin 15[18]。
2.3 多羟基吲哚里西啶(Indolizidine)生物碱
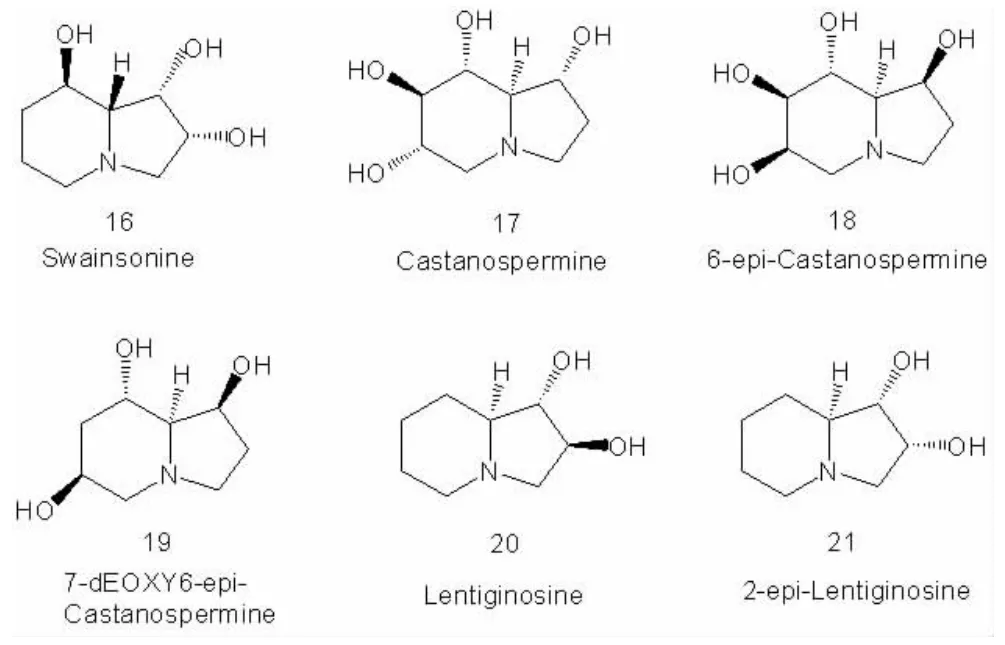
图3 多羟基吲哚里西啶生物碱
随后的1981 年, 人们从澳大利亚板栗树Castanospermum australe的种子中提取出 Castanospermine 17(中文名:栗树精胺),即 1,6,7,8-四羟基-吲哚里西啶生物碱[19]。他是一个双环衍生物,来源于DNJ,其晶体的X-射线衍射数据表明其六员环的立体化学具有葡萄糖的构型[20]。从Castanospermum australe 中还分离得到6-epi-castanospermine 18[21]和7-deoxy-6-epicastanospermine 19, 前者分子中的哌啶环具有D-manno 的构型,它对人体内的中性α-甘露糖苷酶具有好的抑制活性,而后者对真菌的淀粉葡萄糖苷酶具有弱的抑制活性。另两个多羟基吲哚里西啶生物碱2-epi-Lentiginosine 21 与Lentiginosine 20 能够由紫云英Astragalus lentiginous 的叶子中分离得到, 前者对真菌的淀粉葡萄糖苷酶具有相当好的抑制活性,而后者对该酶则没有抑制性[22]。
2.4 多羟基双稠吡咯烷生物碱

图4 多羟基双稠吡咯烷生物碱
随着Alexine 22 从豆科植物Alexa leiopetala 中被分离出来[23],一类全新的C-3 位被碳原子取代的双绸吡咯烷生物碱引起了人们的关注。 之后,人们从Castanospermum australe 的种子中分离到Australine 23[24],其晶体的X-射线衍射数据表明它与Alexine 是互为C-7a 差向异构的一对一非对映异构体, 而且它可被看作是五员环的Castanospermine 或者构象固定的DMDP。Eugneia jambolana(桃金娘属植物)及Casuarina equisetifolia(木麻黄属植物)药用价值极高,从前者的树皮和后者的树叶中,人们均分离到Casuarine 24[25]。 近年来通过分离风信子植物(Scilla campanulata、Hyacinthoids 和 non-scripta),成功得到双稠吡咯烷 Hyacinthacine B1 26 和 Hyacinthacine C1 27,分别替换了 C-3、C-5 上的碳原子[26]。 具有长脂肪链的双稠吡咯烷Broussonetine N 28 是由小构树分离产生,它是第一个双环形式存在的Broussonetines 家族多羟基生物碱[27]。
2.5 多羟基去甲莨菪烷(Nortropane)生物碱
随着Calystegines 的成功分离, 多羟基去甲莨菪烷类生物碱开始作为一个新的种属存在(图17)[28]。 Calystegines 结构特点如下:1)具有N-去甲基莨菪烷的桥环结构骨架;2) 不同位置的桥环共同被多个轻羟取代,形成不同立体结构;3)羟基取代C-1 位(桥头季碳)被羟基最终生成N,O-缩酮。

图5 多羟基去甲莨菪烷生物碱
Calystegines 是人类第一个发现的植物相关性非直接代谢产物,其参与细菌及植物相关联系的调节。 包括Convolvulus arrvernsis 及Calystegia sepium 在内的旋花科植物及包括Physalis alkengi 及Atropa belladonna 在内的茄科植物是Calystegines 的有效载体。 Calystegines还存在于一些可食用的水果和蔬菜(如马铃薯,茄子,番茄,酸浆果和桑葚) 中。 分类上, 现如今流行的分类方法主要是由于不同Calystegines 所含轻基数不同,进而分成的 A 类,B 类和 C 类。 而在这之前其主要分为A,B 两类,分类依据为其在纸电泳中的迁移程度,同时根据HPLC 分离技术进行细化,A 类在此基础上又分为A1,A2,A3等;同样的方法也适用于Calystegine B。 如果Calystegine B 中氨基取代了1 号碳原子上的羟基那么就会形成不同的去甲莨菪烷(Calystegine N), 此外还可以分离获得Calystegines 相关的糖基化产物, 在Nicandra physalodes 果实中, 我们就可以发现葡萄搪糖基化Calystegine B1 3 号碳原子上的羟基后的相关产物[29]。
综上所述,除了少数从微生物中分离外,大多数的多羟基生物碱都是从植物中分离出来的。 从物种演化的角度看,有些微生物或植物产生的多羟基生物碱释放到土壤中而被其它植物吸收并在机体内累积。也有可能某些微生物如根瘤菌,它本身能产生多羟基生物碱,但是由于它与植物共生,后来人们从植物中提取出来,也就理所当然的当作是植物所产生的了。
3 多羟基生物碱与结核病
众所周知,抗结核杆菌主要的作用靶点之一就是其细胞壁,只要能把结核杆菌的细胞壁破坏了,基本上就会消灭结核。 结核杆菌细胞壁主要有阿拉伯呋喃糖、半乳呋喃糖和甘露吡喃糖等物质组成的。 具有抗结核活性的多羟基生物碱类化合物乙胺丁醇(Ethambutol,EMB),其主要的抗结核原理就是能降低阿拉伯呋喃糖糖基转移酶的活性来减少细胞壁中阿拉伯糖的合成的, 使结核杆菌的细胞壁变得十分脆弱,使结核杆菌的存活率降低。

图6 阿拉伯呋喃转换酶抑制剂
阿拉伯呋喃糖转换酶拮抗剂37 和38 是Marotte 等人研制出的抗结核药物[30],这两种药物的结构十分类似,其外在骨架是由多羟基生物碱和阿拉伯呋喃糖苷组成, 多羟基生物碱上的N 被一个或数个阿拉伯呋喃糖烷基化。 药理实验证实:38 对阿拉伯呋喃糖结构酶的抑制作用要远远大于37,其机理尚不清楚。 之后,他们又生成了40、41,它们都是用亚甲基替代了呋喃环或糖苷上的O 来提高其稳定性, 遗憾的是,体外实验表明40 和41 与39 没有明显差别。

图7 UDP-Glaf 转换酶抑制剂
化学家Fleed 等人是最早开始研究抑制半乳呋喃糖相关酶活性药物的, 并终于成功地在1997 年研制出首批降低半乳呋喃糖相关酶活性的多羟基生物碱类药物[31],代表药物就是吡咯烷类多羟基生物碱42 及43, 后来又在深入研究的基础上制成了多羟基生物碱44。 这3种药物的生化结构大致相同,作用也是大致一样,对结核杆菌的杀伤作用却是42<43<44。
牛津大学的相关研究把重点放在了UDP-Glap 转化为UDP-Glaf的过程中,认为只要把这一过程的UDP-Glaf 转换酶加以抑制,就可以达到抑制半乳呋喃糖合成的目的,在这一原则下,在2004 年,Thomas小组合成了UDP-Glap 转化为UDP-Glaf 的中间类似物45,45 可以起到竞争性抑制这一转化过程的作用, 效果较好,45 又分为45a 和45b两个亚型,这两个亚型,虽然组成,结构等都大致相同,但是在抗结核杆菌上,效用大不一样,且45b 几乎没有活性[32]。
2008 年,澳大利亚von Itzstein M 等科研人员以UDP-半乳糖转换酶为靶点,合成了半乳呋喃糖烷基硫代糖苷,发现其具有较好的抗结核活性, 最低抑菌浓度 MIC 是 5mg/ml[33]。 同年, 法国的糖化学家Olivier R.Martin 教授报道了一系列UDP-半乳糖转换酶抑制剂,以UDP-Galf 为底物,模仿其立体构型,设计一系列与其结构类似的目标化合物,经过多步合成反应完成其制备,药理实验尚在进行中[34]。
2009 年,Stéphane P.Vincent 课题组进一步发现了二磷酸尿苷-6-去氧-6-氟-D-半乳呋喃糖可竞争与UDP-半乳糖转换酶结合干扰AG 的合成[35],并证实结构中D-半乳呋喃糖6 位的附近是UDP-半乳糖转换酶与底物相互作用的位点。
4 结束语
经过40 多年的发展, 多羟基生物碱作为小分子量的糖甘酶抑制剂,不仅在糖苷酶的机理研究方面作出了突出的贡献,同时在结核病的治疗方面也给人们带来了福音。正越来越受到学术界以及商业界的广泛关注。 深入展开其结构与活性之间的相关性研究具有重要意义。多羟基生物碱作为一种高效、低毒、无污染、对人畜安全的天然药物必将受到人们的广泛关注。 但由于其天然含量不高且难于分离纯化,因此通过化学手段对其加以修饰或合成, 筛选高活性生物碱开发新药等,都是未来对多羟基生物碱进行进一步研究的热点内容。
[1]ANDRIESK,JOZEFL,MARCELK.Quinolinederivativesforthetreatmentof latenttuberculosis:WO,2006(7048)A1[P].
[2]Qureshi H, Arif A, Alam E, et al.Integration of informal medical practitioners in DOTS implementation to improve case detection rate [J].J Pak Med Assoc, 2010,60(1): 33-37.
[3]Lonnroth K, Castro K G, Chakaya J M, et al.Tuberculosis control and elimination 2010-50: cure, care, and social development [J].The Lancet, 2010,375(9728):1814-1829.
[4]Inouye S,Tsuruoka T, Ito T, et al.Structure and synthesis of nojirimycin[J].Tetrahedron,1968,24(5):2125-2144.
[5]Niwa T, Tsuruoka T, Goi H, et al.Novel glycosidase inhibitors, nojirimycin B and D-mannonic-delta-lactam.Isolation, structure determination and biological property[J].The Journal of antibiotics,1984,37(12):1579-1586.
[6]Holt K E, Leeper F J, Handa S.Synthesis of β-1-homonojirimycin and β-1-homomannojirimycin using the enzyme aldolase [J].J.Chem.Soc., Perkin Trans.1,1994(3):231-234.
[7]Martin O R, Saavedra O M.Concise chemical synthesis of β-homonojirimycin and related compounds[J].Tetrahedron letters,1995,36(6):799-802.
[8]宋婕.桑树资源中1-脱氧野尻霉素的含量测定及桑叶,桑枝和蚕沙中1-脱氧野尻霉素等多羟基生物碱的提取纯化技术研究[D].暨南大学,2011.
[9]Fellows L E, Bell E A, Lynn D G, et al.Isolation and structure of an unusual cyclic amino alditol from a legume [J].Journal of the Chemical Society, Chemical Communications,1979(22):977-978.
[10]Segraves N L, Crews P.A Madagascar sponge Batzella sp.as a source of alkylated iminosugars[J].Journal of natural products,2005,68(1):118-121.
[11]Welter A, Jadot J, Dardenne G, et al.2, 5 -Dihydroxymethyl 3, 4 -dihydroxypyrrolidine dans les feuilles de Derris elliptica [J].Phytochemistry, 1976,15(5):747-749.
[12]Bautista M, Andres D, Cascales M, et al.Effect of gadolinium chloride on liver regeneration following thioacetamide-induced necrosis in rats[J].International journal of molecular sciences,2010,11(11):4426-4440.
[13]Asano N, Oseki K, Tomioka E, et al.N-containing sugars from Morus alba and their glycosidase inhibitory activities [J].Carbohydrate research,1994,259(2):243-255.
[14]Watson A A, Nash R J, Wormald M R, et al.Glycosidase -inhibiting pyrrolidine alkaloids from Hyacinthoides non-scripta[J].Phytochemistry,1997,46(2):255-259.
[15]Li Y X,Huang M H, Yamashita Y, et al.L-DMDP, L-homoDMDP and their C-3 fluorinated derivatives: synthesis and glycosidase-inhibition [J].Organic &biomolecular chemistry, 2011, 9(9):3405-3414.
[16]Shibano M, Tsukamoto D, Masuda A, et al.Two new pyrrolidine alkaloids,radicamines A and B, as inhibitors of alpha-glucosidase from Lobelia chinensis Lour[J].Chemical & pharmaceutical bulletin,2001, 49(10): 1362-1365.
[17]Shankaraiah G, Sateesh Chandra Kumar R, Poornima B, et al.Stereoselective synthesis of (+)-radicamine B[J].Tetrahedron Letters, 2011,52(38):4885-4887.
[18]Parker A J.Solvation of ions—enthalpies, entropies and free energies of transfer[J].Electrochimica Acta, 1976, 21(9): 671-679.
[19]吴达,师彦平,梁冰,等.苦马豆素研究进展[J].中草药,2003,34(4): 5-7.吴旭锦,杨鸣琦,白春黎,等.苦马豆素的来源及分离方法进展[J].动物医学进展,2005, 26(5):41-44.
[20]Allan G, Ouadid -Ahidouch H, Sanchez -Fernandez E M, et al.New Castanospermine Glycoside Analogues Inhibit Breast Cancer Cell Proliferation and Induce Apoptosis without Affecting Normal Cells[J].PloS one, 2013, 8(10): e76411.
[21]Yun H, Kim J, Sim J, et al.Asymmetric Syntheses of 1-Deoxy-6, 8a-di-epicastanospermine and 1-Deoxy-6-epi-castanospermine [J].The Journal of organic chemistry, 2012,77(12): 5389-5393.
[22]Zhuang J J, Ye J L, Zhang H K, et al.An unexpected high erythro-selection in the Grignard reaction with an N,O-acetal: a concise asymmetric synthesis of indolizidine alkaloid(?)-2-epi-lentiginosine[J].Tetrahedron,2012,68(6):1750-1755.
[23]Nash R J, Thomas P I, Waigh R D, et al.Casuarine: a very highly oxygenated pyrrolizidine alkaloid[J].Tetrahedron letters,1994,35(42):7849-7852.
[24]Gilles P, Py S.SmI2-Mediated Cross-Coupling of Nitrones with β -Silyl Acrylates: Synthesis of (+)-Australine[J].Organic letters,2012,14(4):1042-1045.
[25]Parmeggiani C, Cardona F, Giusti L, et al.Stereocomplementary Routes to Hydroxylated Nitrogen Heterocycles: Total Syntheses of Casuarine, Australine, and 7‐epi‐Australine[J].Chemistry-A European Journal,2013,19(32):10595-10604.
[26]Reddy P V, Smith J, Kamath A, et al.Asymmetric Approach to Hyacinthacines B1 and B2 [J].The Journal of organic chemistry, 2013, 78(10):4840-4849.
[27]ShibanoM,TsukamotoD,KusanoG.Anewpyrrolizidinealkaloid,Broussonetine N,asaninhibitorofglycosidase,from BroussonetiakazinokiSieb.andabsolute stereostructuresofBroussonetinesAandB[J],1999,47:907-908.
[28]KatoA,AdachiI,MiyauchiM,etal.Polyhydroxylated pyrrolidineand pyrrolizidinealkaloidsfrom Hyacinthoidesnon-scriptaandScillacampanulata[J].Carbohydrateresearch,1999,316(1):95-103.
[29]GriffithsRC,WatsonAA,KizuH,etal.TheisolationfromNicandraphysalodes and identification of the 3-O-β-D-glucopyranoside of 1α,2β,3α,6α-tetrahydroxy-nor-tropane(CalystegineB1)[J].Tetrahedronletters,1996,37(18):3207-3208.
[30]ReynoldsRC,BansalN,RoseJ,etal.Ethambutol-sugarhybridsaspotential inhibitorsofmycobacterialcell-wallbiosynthesis[J].Carbohydrateresearch,1999,317(1):164-179.
[31]ChaumontetM,PonsV,MarotteK,etal.Hydrolyticallystable arabinofuranosideanalogsforthesynthesisofarabinosyltransferaseinhibitors[J].Tetrahedronletters,2006,47(7):1113-1116.
[32]MeijerMD,RumpM,GossageRA,etal.New “bucky-ligands”.Potentially monoanionicterdentatediaminoarylpincerligandsanchoredto C[J].Tetrahedron letters,1998,39(37):6773-6776.
[33]Davis C B, Hartnell R D, Madge P D, et al.Synthesis and biological evaluation of galactofuranosyl alkyl thioglycosides as inhibitors of mycobacteria[J].Carbohydrate research,2007,342(12):1773-1780.
[34]Liautard V, Desvergnes V, Itoh K, et al.Convergent and Stereoselective Synthesis of Iminosugar-Containing Gal f and UDP-Gal f Mimicks: Evaluation as Inhibitors of UDP-Gal Mutase [J].The Journal of organic chemistry,2008,73(8):3103-3115.
[35]Eppe G, Peltier P, Daniellou R, et al.Probing UDP-galactopyranose mutase binding pocket: a dramatic effect on substitution of the 6-position of UDP-galactofuranose[J].Bioorganic & medicinal chemistry letters, 2009,19(3):814-816.
猜你喜欢
健康体检与管理(2022年2期)2022-04-15
化工时刊(2020年4期)2020-06-07
中成药(2018年5期)2018-06-06
中成药(2017年8期)2017-11-22
化工管理(2017年26期)2017-03-04
合成化学(2016年5期)2016-06-13
合成化学(2015年1期)2016-01-17
分析测试学报(2015年5期)2016-01-13
中国药理学通报(2014年2期)2014-05-09
天然产物研究与开发(2014年3期)2014-04-27
