
核酸中碱基的氢键作用机理及电子特征理论研究
2014-03-07 03:48刘玉震马飞燕冀利妃
化学研究 2014年2期
肖 祎,方 意,刘玉震,马飞燕,冀利妃
(西南大学化学化工学院,重庆 400715)
核酸中碱基的氢键作用机理及电子特征理论研究
肖 祎*,方 意,刘玉震,马飞燕,冀利妃
(西南大学化学化工学院,重庆 400715)
采用耦合簇量子化学方法CCSD/aug-cc-pVDZ研究了嘧啶与嘌呤之间的相互作用,利用基函数叠加误差法(BSSE)消除相互作用能误差,并进行了几何结构优化;采用Gaussian 03程序包中的NBO程序分析了二阶稳定化能及自然键轨道.与此同时,应用约化密度函数(RDG)填色等值面图对体系进行了图形化分析,分析了氢键相互作用所在的空间位置和相对强度,以及氢键相互作用的性质,以进一步了解二者的相互作用.结果表明,嘧啶-嘌呤体系的相互作用属于闭合壳层静电相互作用.电子密度跃迁矩阵分析结果表明,激发区域主要集中在N原子和O原子处,涉及的空间广度很大,第一激发态主要涉及前线分子轨道,属于σ→π*或n→π*类型跃迁.
核酸;碱基;氢键作用机理;电子特征;理论研究
1953年WATSON和CRICK发现了DNA双螺旋结构[1-2],两条多聚核苷酸链相互反平行盘绕成双螺旋;互补碱基由位于螺旋内部的氢键联结,其平面垂直于螺旋轴.碱基对通过氢键作用形成一定的空间结构,此种结构具有调节基因表达的生物功能,与动植物生长发育、疾病发生有着密切关系.有了碱基间氢键[3-4]的作用才能构成丰富复杂的DNA分子,如果没有碱基之间的氢键,就不可能组装成DNA双螺旋链;因此核碱基氢键的研究越来越受到科学工作者的重视.氢键无处不在,是自然界中很重要的相互作用方式,它已渗透到化学、生物、物理以及材料科学等[5-7]领域.维持DNA双螺旋链主要是互补碱基间的氢键、碱基堆积力[8]等,氢键是维系和促进蛋白质与核酸高级结构形成的重要作用力,基因突变损伤时氢键的变化与基因的特殊生物学行为密切相关.大连化学物理研究所的霍红等[9]曾对乳腺癌组织中蛋白质与核酸分子氢键进行了红外光谱研究,癌组织具有明显而有规律的光谱特征,因此有可能利用氢键的光谱特征作为诊断和预测突变可能性的指标,在生命科学上有着重要意义.
核酸碱基中,主要存在N-H…N及N-H…O两种强氢键类型,静电引力导致分子间超共轭效应起主要作用,而分子内共轭及原子杂化效应都可以忽略.ALABUGIN[10]理论能够很好的解释氢键位移的形成,氢键的红移和蓝移的形成原因主要是分子间超共轭效应和原子轨道重杂化两种效应共同决定的,当超共轭效应大于重杂化效应时形成红移氢键,反之则形成蓝移氢键.分子间和分子内超共轭能大小是其效应强弱的具体体现.NBO分析分子间和分子内共轭二阶稳定化能有助于对氢键形成机理进行解释.HOBZA等[11]提出用两步机理理论解释蓝移氢键的形成,即电荷转移到电子受体的σ*(N-H)反键轨道或具有孤对电子原子上,导致质子给体原子内部结构发生重排,电荷分布均增大从而导致X-H键伸长、频率减小.本文作者主要采用上述作用机理对σ*(N-H)作为质子给体与n(N、O)作为质子受体形成的红移氢键作用机理进行解释.
1 计算方法
所有单体和复合物结构均采用耦合簇(CCSD)方法[12]结合aug-cc-pVDZ基组进行了全优化,并在优化的基础上做频率分析.CCSD方法证实适用于氢键相互作用的研究[13],能量经基函数叠加误差法(BSSE)[14]消除误差,得到校正后相互作用能ΔEBSSE,所有计算都采用Gaussian 03[15]程序包完成.其后采用该程序包中的NBO程序对其进行自然键轨道分析.应用约化密度梯度(RDG)[16]填色等值面图对体系进行了图形化分析,RDG填色等值图使用VMD程序绘制,在优化好的复合物结构中,各方向格点数为80,80,80来计算空间内各点的RDG函数和sign[λ2(r)]ρ(r)函数的值;此外采用AIM[17]方法分析氢键的成键路径和键临界点,通过分析键的临界点的拓扑性质,讨论电子密度、拉普拉斯量与氢键强弱的关系,从而说明碱基间确实形成了氢键.
2 结果与讨论
2.1 几何构型、频率及相互作用能
所有复合物的优化构型示于图1,红色数字为氢键键长(nm),绿色为键角(°).具有孤对电子的n(N)和n(O)作为质子受体形成的N…H-N、O…H-N键角均在175°~179°之间(趋于直线型),而较弱的O…H-C氢键的键角为132.34°.表1列出了嘧啶与嘌呤形成的未校正(ΔE)和BSSE校正过的相互作用能(ΔEBSSE).未校正的相互作用能与校正过的相互作用能均为负值,说明形成的复合物为稳定构型.鸟嘌呤与胞嘧啶相互作用能的绝对值最大,说明其最为稳定.由于腺嘌呤是尿嘧啶的甲基化衍生物,它们具有类似的分子结构,所以A-T与A-U的稳定性相近.在A-T与A-U中由于存在三个不等价氢原子,所以形成了N-H…N、N-H…O和C-H…O三种氢键.由于C-H…O键长较长、键角很小,属于弯曲型的蓝移氢键,它们对体系的稳定性贡献小,本文可以忽略其影响.综合分析碱基间只存在两种较强氢键形式:N-H…N和N-H…O,A-T及A-U的两个氢键结构使碱基间形成了一个类似八元环平面结构;同时鸟嘌呤与胞嘧啶形成了N…H-N及两个N-H…O氢键,三个氢键结构使碱基间形成了两个类似八元环的结构.键的对称性及极化作用使N…H-N强于O…H-N,理论上氢键对复合物稳定性贡献顺序:N…H-N>O…H-N>>O…H-C.
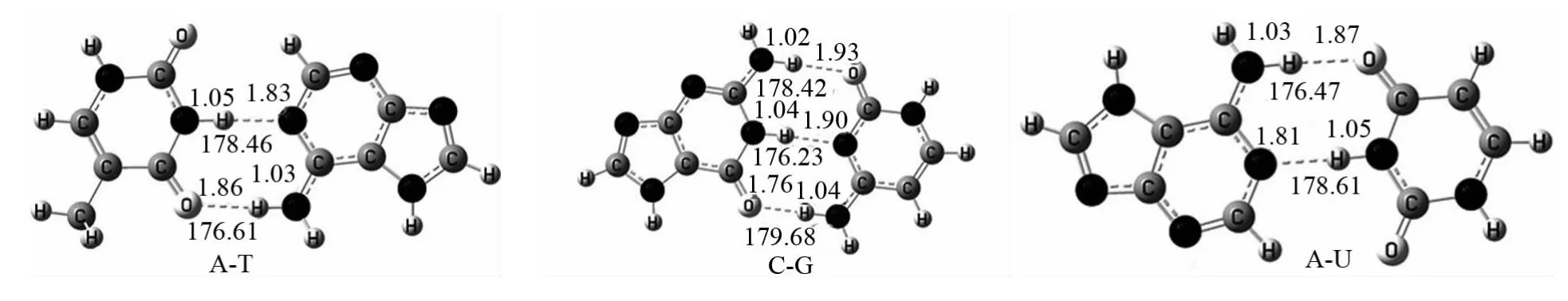
图1 几何结构及相关结构参数Fig.1 Geometric structure and the relate structural parameters
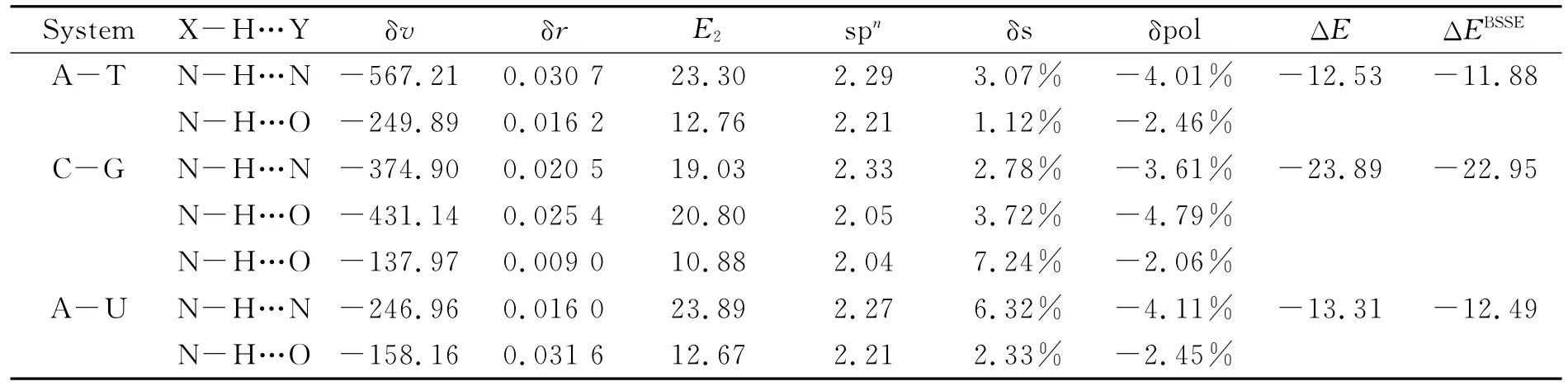
表1 结构参数变化,能量分析以及NBO分析Table 1 Structural parameter change,energy and NBO analysis
从表1可知,相互作用能ΔE的大小往往与复合物的几何结构紧密相联,与单体相比,N-H键均出现伸长、振动频率减小,属于传统的红移类型氢键.在这些红移氢键中,N-H键长伸长明显,δr普遍大于0.016 0nm,频率改变也很大,δv大于100cm-1.频率红移程度与前面给出的相互作用能相关,从计算的结果看,三个碱基对的相互作用能分别为:-12.53,-23.89,-13.31kcal/mol.NBO理论可以从轨道相互作用的角度来理解氢键的形成机理,同时也可清楚地了解每个原子的电荷变化及原子轨道杂化情况δs,N-H键的氮原子s轨道成分均明显增大(1.12%~7.24%),说明存在较大的原子重杂化效应,同时键的极化(δpol)值(-4.79%~-2.06%)为较大的负值,说明存在很大的负极化效应;最终N-H键在较强的正重杂化效应和负的极化效应共同作用下出现明显的红移现象.碱基环本身可以看成较大的π电子体系,所以形成的主要是π型氢键,电子离域程度越大形成的复合物稳定性越大.相互作用能大小与氢键的类型和形成数目密切相关,键越强且数目越多体系就越稳定.由于氢键O…H-C的键能比O…H-N与N…H-N小得多,属于弱氢键.氢键增强了碱基在DNA双螺旋结构中的稳定性,促使碱基更趋向于稳定型构型配对,在一定程度上避免了碱基错配、DNA突变的发生.
ALABUGIN等[10]指出氢键出现红移或蓝移均可以使用超共轭和重杂化两个概念来解释.超共轭效应会使N-H键伸长,而轨道重杂化效应则会使N-H键长缩短,两种效应共同作用决定了N-H键长的改变和频率的位移特征.电荷从含有孤对电子的成键轨道转移到σ*(N-H)反键轨道上,引起电子受体轨道电荷发生重排,分子间超共轭作用导致电荷转移伸长效应大于分子内共轭及轨道杂化收缩效应.N原子与相邻C原子间s轨道覆盖增大导致N-C键收缩,因此氢键导致质子给体N-H先收缩再伸长.而O…H-C氢键由于分子内共轭效应导致C与相邻的N价层s轨道特性覆盖减小,C-N键级减小而不足以消去C轨道杂化收缩效应,分子间超共轭效应被分子内共轭效应削弱而导致键长收缩,电荷分布不均、键级减小,出现蓝移现象.
2.2 氢键的电子密度拓扑分析
BADER提出的AIM理论通过研究临界点的电子密度拓扑性质,把分子的性质与构成它的原子的性质联系了起来,并且给出了氢键的定义.POPELIE[18]在AIM理论基础上提出了氢键判断的标准:在H原子与质子接受原子体Y之间存在临界点(BCP)与相应的成键路径,两原子都有各自的特征原子区域且相互穿透;氢键的BCP处的电荷密度在0.002~0.034a.u.之间,电子密度的拉普拉斯值▽2ρ在0.024~0.139a.u.之间,H原子净电荷增大,能量增大,体积减小.根据这个标准对体系的氢键进行深入的研究,做出化学键的直观的物理图像,达到定量地描述化学键的性质及强度的目的.临界点处电子密度ρ(rc)的拓扑值与化学键的性质密切相关.一般键临界点处的电子密度越大,则两个原子之间的化学键越强,反之则越弱.因为ρ(N…H-N)>ρ(O…H-N)>>ρ(C-H…O),从而可知三个氢键的强弱顺序:N…H-N>O…H-N>C-H…O.
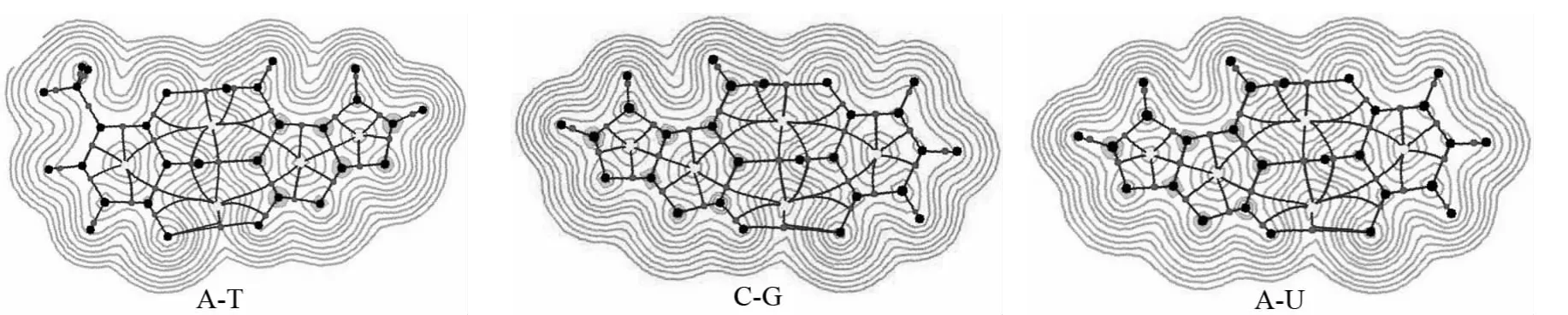
图2 电子电荷密度等值线图Fig.2 Electron charge density contour map
为了更深入地了解形成氢键的本质,我们对所有复合物进行了电子密度拓扑分析,图2给出了复合物的电子密度等值线图,图中红色圆点为平面内与面外的(3,-1)键临界点,黄色圆点为作图平面内与面外的(3,+1)环临界点.根据电子密度拓扑分析结果,在N-H…Y之间均有键临界点,表明存在Y…H-N氢键.随着氢键的变长,键临界点电子密度减小,N-H电子密度ρ(N-H)有所减小而与其相邻的C-N键则有所增大.一般来说,在形成氢键时,质子给体中主要是反键轨道受影响,成键轨道的电荷占据数变化不大.对于氢键,ρ(N-H)减小表明σ*(N-H)电荷占据增大,因此质子给体的部分电子密度转移到相邻的σ*(CN)上;我们计算发现Y原子域中的电子密度增大,表明存在从质子给体到质子受体的电子转移.
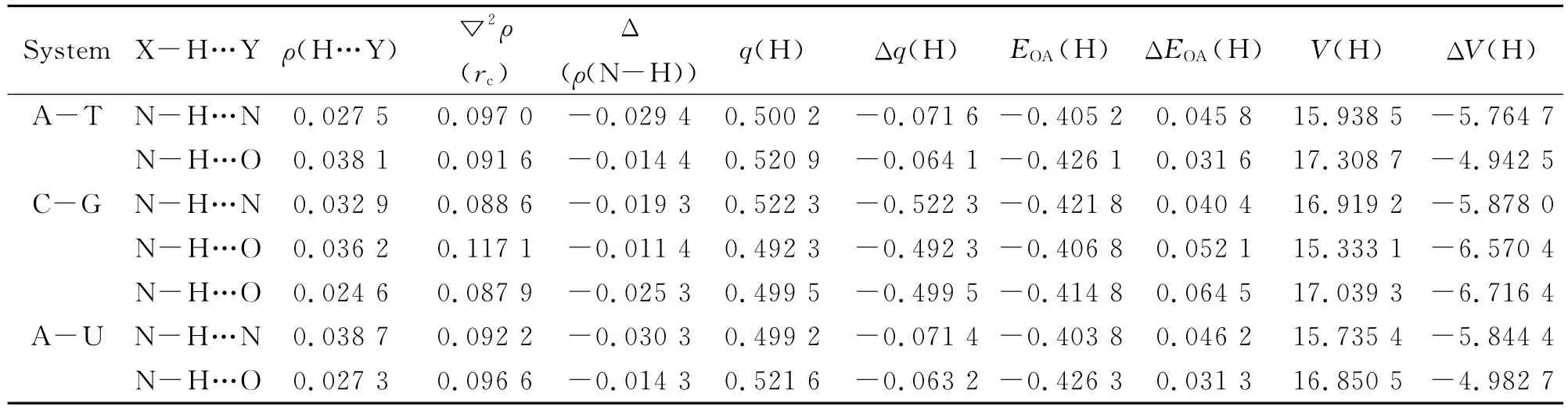
表2 氢键键临界点及H原子的拓扑性质分析(a.u.)Table 2 The topological properties analysis of the hydrogen bond at the bond critical point and H atom(a.u.)
按BADER的观点,键临界点处拉普拉斯量▽2ρ(rc)标志着化学键的特性.表2数据显示,H…Y间键临界点的电子密度ρ在0.024 6~0.038 7a.u.之间,▽2ρ(rc)值在0.087 9~0.117 1a.u.之间,拉普拉斯值为正,表明氢相互作用是闭壳层作用.由于氢键,质子供体与受体的键临界点电子密度ρ(N-H)和ρ(Y)都有一定程度增大(后者更大).H和Y原子相互渗透导致H均表现出电荷减小、能量增大及体积减小特征.成键后N和Y(N、O)的体积出现了明显的收缩趋势,强吸电子作用使其电子云密度增大导致原子体积缩小.总体来说供体原子体积均有明显减小,因氢键的类型不同而有所差异,整体变化趋势与成键越强收缩越明显一致.此外分析分子间两个环临界点即(3,+1),此点是势能面上的鞍点,环临界点的电子密度范围为0.004 8~0.005 2a.u.,电子密度非常分散,分子间环的形成在一定程度上增强了氢键键合物的稳定性,使DNA双螺旋结构更稳定.
2.3 表面静电势分析
图3列出了在CCSD/aug-cc-pVDZ水平下嘌呤和嘧啶的静电势[18]图,图中用不同的颜色标记了不同区域静电势的大小,其中红色表示具有较大的负的电势,蓝色表示具有较大的正的电势,其他颜色如绿色和黄色等表示处于中间的电势.静电势由原子核与电子共同贡献,原子核贡献正值,电子贡献负值.在成键区域、原子壳层区域,电子是聚集的,但由于离原子核太近,仍然基本是正值.而孤对电子区域,因电子富集且空间范围弥散,离原子核有一定距离,所以能呈现负的静电势.嘌呤和嘧啶的杂原子(N,O)附近都具有较大的负电势,故能与其互补碱基的σ*(N-H)等具有较大的正电势的分子形成N-H…Y氢键.根据图3可知在氢原子附近的分子表面上都存在静电势极大点,这是因为氢的电负性小于碳、氧、氮,因此氢原子都显正静电势.由于氮原子比碳原子电负性强,故负的静电势更显著,图示中有些氢原子附近不像其他氢原子那样有对应的极大点,这是因为相邻N-H键的氢原子的正电性太强了,造成其附近分子表面上的静电势较大,削弱其附近区域负的静电势的贡献,因此H较弱的正电性对静电势的影响不明显,无法形成极大点;而出现的极小点是由于互相靠近的N、O孤对电子密集的电子云产生的.A-T中1号和2号极小点静电势值相近且空间效应相似,体现的是π-π电子云对静电势的显著负贡献.从这两个极小点位置和原子涵盖的区域可以看出,在N-H对位原子附近分子表面的静电势比在碳原子附近更负,因此更容易被亲电试剂所进攻,是邻对位取代基;这也在一定程度上解释了DNA中戊糖C原子优先在此部位与N成键的原因.
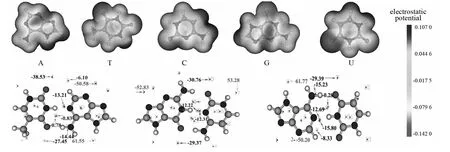
图3 碱基单体静电势和碱基对表面静电势极值点分布图Fig.3 Base monomers electrostatic potential and base-pairs extreme value point of surface electrostatic potential distribution
图3中的极值图中绿色是极大点,粉色是极小点,而蓝色数字代表极大点极值,红色数字代表极小点极值.由分析可知,N-H中氢对应表面静电势极大点,而N原子对应极小点,因此这两个原子最容易靠静电相互作用结合到一起,并且是极大点正冲着极小点的这种朝向,这可以使复合物能量最大程度降低,这也间接说明了常规氢键的本质在很大程度上可以解释为静电相互作用.极小点基本上体现的是氮氧等的孤对电子对静电势的负贡献.而在相邻的两个β碳(邻位碳)正上方,体现的是π电子对静电势的负贡献.有些极大点静电势数值为负,化学意义不大,可无视之.氮氧原子附近的分子表面静电势最负,这是因为其孤对电子对静电势有很大的负贡献.由于其附近分子表面上没有正值区域,所以正值区域的静电势值显示的是较小负值.碱基中碳原子裸露在分子表面上的区域主要是体现π电子特征的区域,由于π电子云使这部分区域静电势为负,所以碳原子附近静电势平均值也都为明显负值,并且无正值区域.这种讨论分子表面上对应不同原子区域的定量数据的方法和分子表面极值点分析往往会得到共同的结论.通过分析分子对应的分子表面上的局部区域的静电势平均值等数据就可以定量考察其特征[19].
2.4 跃迁密度矩阵及前线分子轨道分析
已知两个电子态G、E的波函数,就可以构建它们之间的跃迁密度矩阵函数:

其中N是总电子数,x是自旋空间坐标,s是自旋坐标,r是空间坐标.跃迁密度矩阵的对角元,代表电子态跃迁造成的原子电荷变化的程度;而非对角元代表的是电子跃迁时原子与相邻原子的电子-空穴的相干程度.以原子编号作为横、纵坐标,以颜色来表示数值大小绘制跃迁密度矩阵图,可以很方便地分析电子跃迁所涉及到的原子以及原子间相干范围,尤其是对于较大的共轭分子来说,尤为明显.本文采用半经验的ZINDO=1方法计算碱基对基态S0到第一激发态S1的能级跃迁,同时结合含时密度泛函(TD-DFT)方法研究碱基对的垂直激发能(TD-DFT方法近几年被广泛证明适用于计算分子的能级光谱特性[20]).
从图4可见,激发区域比较强烈的部分均集中在图的对角区域,说明这种激发模式主要涉及的是嘧啶和嘌呤环的N、O等原子的局部跃迁,氢键属于分子间弱相互作用,激发很难跨越它而涉及碱基对的整个分子跃迁.对角线区域是激发强度范围整体较强的区域,其长度体现了电子激发涉及的空间广度很大.图中在非对角元上也有一定的数值,这说明在这种激发模式中原子与邻近原子有较强的电子-空穴相干性.图中非对角描述的带状区域越宽,代表原子间相干区域越大.显然碱基对的跃迁几乎对应的都是涉及分子的全局跃迁,由于碱基环具有很强的电子离域特性,不难理解在这些跃迁中分子内原子间有明显相干性.环内相邻原子在跃迁中相干性较弱,这主要是由于碱基对的结构扭曲破坏了环与周围部分原子间的电子离域性.

图4 碱基对基态到第一激发态的跃迁密度矩阵图Fig.4 Transition density matrix figures of base-pairs from ground state to the first excited state
分子间相互作用过程中最先起作用的分子轨道通常是最高占据轨道HOMO,这是因为分子的HOMO对其电子的束缚较为松弛,具有电子给予体的性质,而最低空轨道LUMO则对电子的亲和力较强,具有电子结合体的性质,这两种轨道最易相互作用.氢键属于弱分子间相互作用,激发过程也容易在此产生,因此通过分析其激发波长可以判断相互结合的强度.在作用过程中起极其重要的作用.氧原子p轨道孤对电子与碱基环的π电子轨道接近平行,能够离域形成一个大π键;而C-G的C=O键与环π键趋向于垂直,导致离域效应降低.整体可以看出HOMO轨道主要体现在嘌呤环,而LUMO则体现在嘧啶环.A-T的能级差为0.148 7a.u.,大于C-G的0.139 6a.u.(图5),再次表明C-G的结合趋势大于A-T.
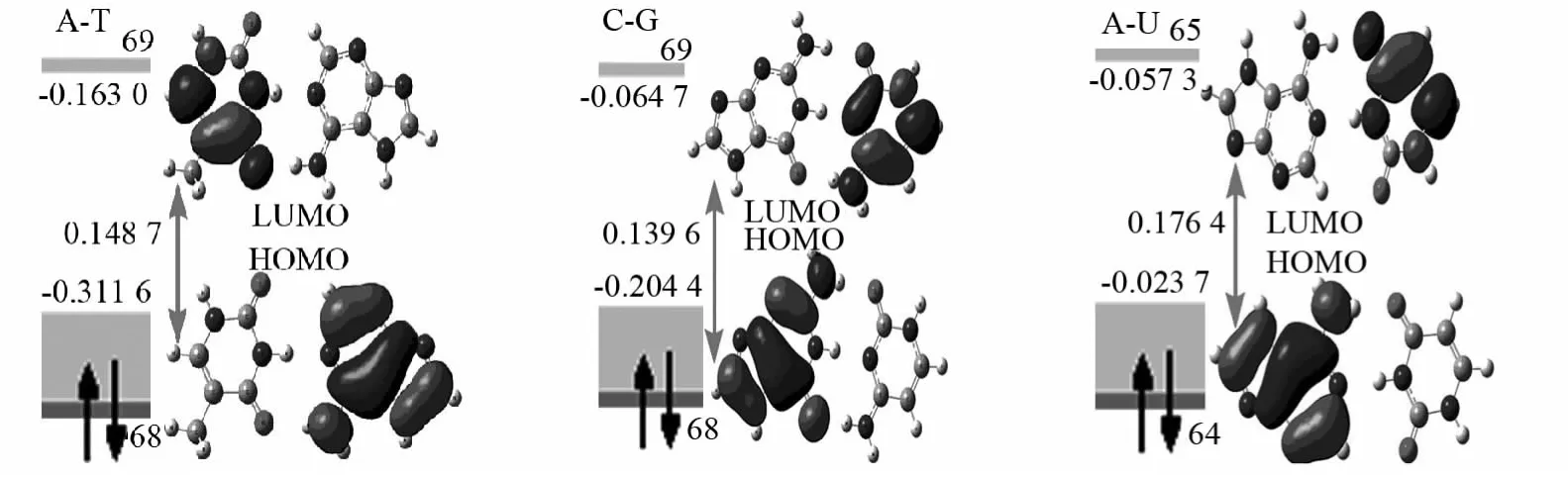
图5 分子前线轨道HOMO与LUMO作用能级(a.u.)示意图Fig.5 Molecular frontier orbital HOMO and LUMO energy levels(a.u.)diagram
采用TD-B3LYP/aug-cc-pVDZ方法对碱基基态S0进行单重态垂直激发能的计算,得到电子吸收波长(λ)及振荡强度(f).从表3可以看出A-T的S0→S1电子吸收波长为281nm,而C-G和A-U的分别在368和287nm处,结合前文分析可知该激发波长与碱基对的稳定性是一致的(C-G>A-T>A-U),即稳定性越大电子吸收波长越大.从表3还可以看出第一激发态主要源于HOMO到LUMO的跃迁,图5给出了前线分子轨道图.
由图5可知,HOMO轨道主要体现在嘌呤环,而LUMO则体现在嘧啶环,因此该跃迁属于分子间电荷转移跃迁机理(CT).由于氢键是弱键,相对于分子内化学键非常弱,因此分子间的部位也最容易发生激发;而其他两个激发带由于涉及非前线最高占据轨道或最低空轨道,跃迁前后的轨道几乎都在嘌呤环上或者都在嘧啶环上,出现分子内电荷转移,所以属于分子内电荷转移机理(ICT).激发时分子的共轭链延长,电荷更分散,发生跃迁后电子的离域程度增大.从基态到第一激发态的跃迁是碱基间的σ→π*或n→π*类型轨道跃迁,而从基态到第二及以上的激发态的跃迁由于涉及非前线轨道,同时存在原子s轨道重杂化效应,所以属于π→π*型轨道跃迁.由于氢键作用位点的不同而具有明显不同的电子吸收波长.由表3可知,他们的电子吸收光谱均在近紫外区域,均出现三个不同波长的吸收谱带,谱带振荡强度均较小,属于禁阻跃迁或半禁阻跃迁.
2.5 RDG函数等值面图形化研究弱相互作用
杨伟涛课题组[21]提出了一种新的可视化研究弱相互作用的方法,此方法的主要目的在于凸显出体系中涉及弱相互作用的区域,以便直观地了解到分子中哪些区域与弱相互作用有关.Multiwfn 在网格设置中提供了一个选项方便研究局部弱相互作用的功能,将弱相互作用在其临界点处的电子密度ρ(r)和sign(λ2)函数相乘而得的sign(λ2)ρ(r)函数投影到RDG等值面上,则弱相互作用的位置、强度、类型都能清晰地显现出来(图6).图中色彩刻度设定为蓝>绿>红,色彩刻度设为[-0.04,0.02],蓝色区域表明ρ(r)较大、sign(λ2)=-1,表现的是较强、起吸引作用的弱相互作用,符合这个特征最常见的就是氢键.绿色区域表明ρ(r)很小,说明相互作用强度很弱,范德华作用区域符合这个特征.红色区域ρ(r)较大、sign(λ2)=+1,对应于在环、笼中出现的较强的位阻效应区域,产生张力,因而红色等值面周围原子间起互斥效应.
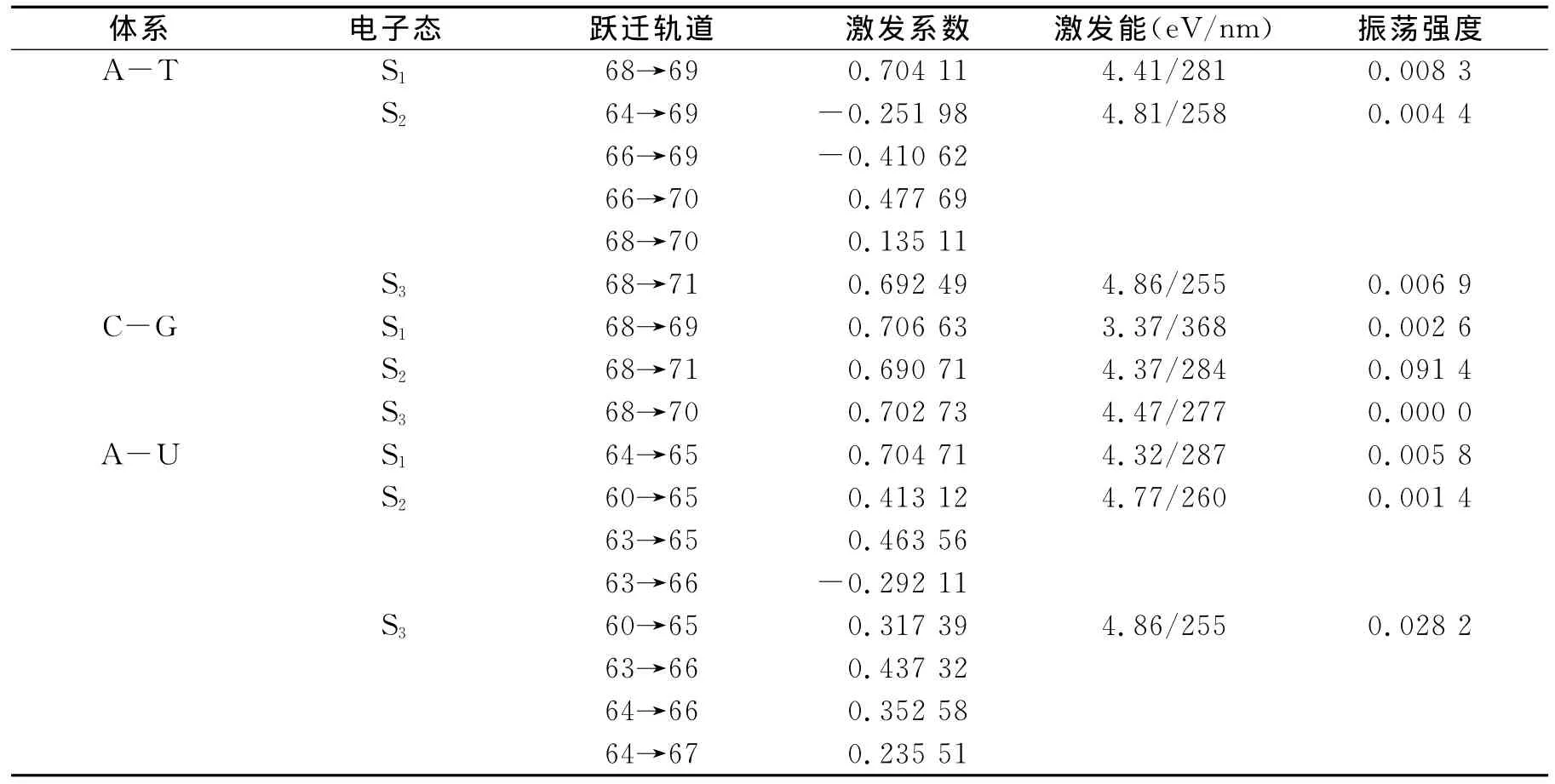
表3 TD-DFT碱基对垂直激发能及振荡强度Table 3 TD-DFT vertical excitation energies and oscillator strength for base-pairs
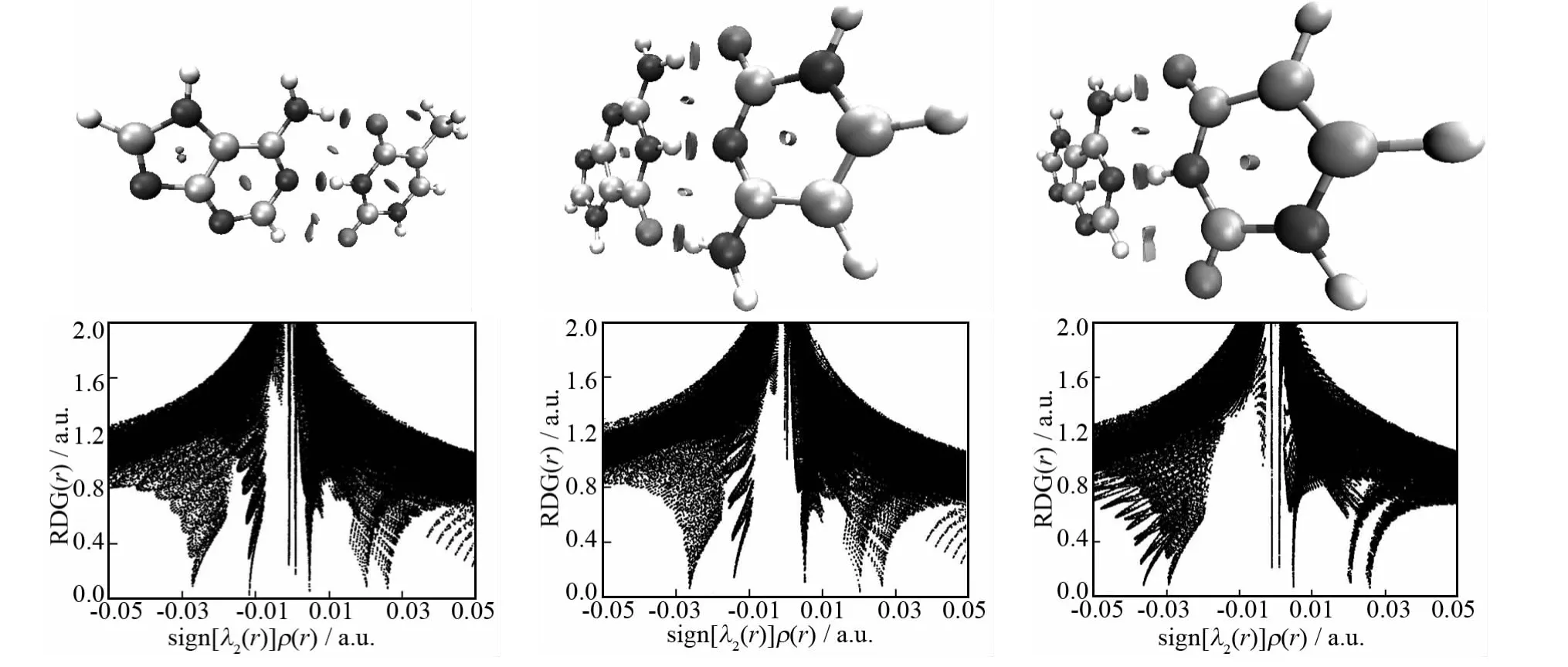
图6 碱基对的约化密度梯度函数(RDG)示意图(等值面:0.8)(上)和散点图(下)Fig.6 Base pairs of reduced density gradient function(RDG)diagram(isosurface:0.8)(top)and scatter diagram(bottom)
约化密度梯度函数散点图中,sign[λ2(r)]ρ(r)函数的范围定义在-0.05~0.05a.u.之间,其中蓝色越深说明相互作用越强,而接近红色的部分说明排斥作用大(如空间位阻效应越大),绿色部分体现范德华相互作用.RDG对应sign[λ2(r)]ρ(r)散点图中存在一条垂直于sign[λ2(r)]ρ(r)数值的“突刺”,且“突刺”垂直的sign[λ2(r)]ρ(r)数值越小,相互作用越大[23].散点图中左侧“突刺”sign[λ2(r)]ρ(r)<0则存在氢键相互作用以及范德华相互作用,右侧“突刺”是sign[λ2(r)]ρ(r)>0则存在分子间排斥作用.RDG填色等值面图很清晰地显示出了氢键相互作用的具体位置.图中复合物都有蓝、绿、红三种颜色,其中复合物C-G比复合物A-T和A-U中蓝色的填色区域都要深,说明复合物C-G相互作用最大.三个复合物的蓝色区域周边存在绿色部分,表明该区域同时存在较弱的范德华相互作用.此外等值面图的绿色和蓝色中还含着红色,说明碱基对成键形式不能从任何方向靠得太近,否则会产生较大斥力.只有当吸引作用能克服斥力时,体系才能在位阻效应存在的情况下保持稳定.散点图左边的突刺的sign[λ2(r)]ρ(r)很负,对应很强的氢键,因而相应的等值面为蓝色.散点图上突刺并不致密的部分,说明落在这个空间区域的格点较少.右侧的突刺的sign[λ2(r)]ρ(r)为较小正值,对应于图中棕色等值面,体现的是微弱的位阻效应.比较可知,靠弱相互作用结合的复合物,即使存在位阻效应也不会太强,否则将不足以被弱相互作用抵消掉.在碱基环中间有红色梭形区域,体现较强位阻效应,对应散点图最右边的突刺.氮和氧原子与氢原子之间的RDG等值面一小半是橘红色,一大半是天蓝色,这个等值面说明原子间既存在着位阻效应,也存在着弱氢键作用,互斥和吸引效应并存.
3 结论
采用耦合簇方法对核酸中碱基的氢键体系进行构型优化、能量分析、电子密度的拓扑性质及图形化处理,超共轭和重杂化两个概念可以解释电荷转移效应会使N-H键伸长、频率减小,而杂化收缩效应导致N-H键缩短.RDG图形化分析揭示了氢键相互作用所在的空间位置以及相对强度,散点图则说明当吸引作用能克服排斥效应时,体系才能保持稳定.分子间相互作用过程中最先起作用的分子轨道就是前线分子轨道,电子密度跃迁分析表明第一激发态主要源于分子中HOMO、LUMO间跃迁,涉及的是n→π*或π→π*型轨道跃迁,属于分子间电荷转移跃迁机理(CT),其他谱带由于非前线轨道参与作用,原子p轨道出现重杂化作用,属于σ→π*型轨道跃迁的分子内电荷转移机理.碱基的氢键的变化必然引起他们的电子特征发生改变,从而可以作为诊断和预测某些疾病的可能性的指标.
本文的电子版可以从网站http://hxya.cbpt.cnki.net下载.
[1]向义和.DNA纤维的X射线衍射分析与双螺旋结构的发现[J].大学物理,2005,24(1):50-58.
[2]BANDYOPADHYAY D,BHATTACHARYYA D.Estimation of strength in different extra Watson-Crick hydrogen bonds in DNA double helices through quantum chemical studies[J].Biopolymers,2006,83(3):313-325.
[3]GEFFREY G A.Hydrogen bonding in biological structures spring-verlay berlin hoidel-berg[M].New York,1995,1-5.
[4]冯丰,于建国,方维海,等.DNA和RNA双链稳定性差异的理论研究[J].高等学校化学学报,2009,30(12):2445-2451.
[5]DESIRAJU G R.The design of organic solids[M].Amsterdam:Crystal Engineering Press,1989.
[6]CHAKRABORTY S,DUBEY R,JOSEPH S,et al.Crystal engineering in the Desiraju research group in Bangalore[J].Crystalgrowth des,2012,12(10):4688-4691.
[7]DESIRAJU G R.The weak hydrogen bond in structural chemistry and biology[M].Oxford University Press:New York,USA,1997.
[8]卢龙斗,孙富丛,邓传良,等.维持DNA结构稳定性的因素[J].生物学通报,2012,47(6):12-13.
[9]霍红,王幸福,车迅,等.乳腺癌组织中蛋白质与核酸分子氢键特征的研究[J].光谱学与光谱分析,2001,21(5):614-616.
[10]ALABUGINl I V,BONUS,N.Selective transition state stabilization via hyperconjugative and conjugative assistance:Stereoelectronic concept for copper-free click chemistry[J].J Org Chem,2012,77(1):75-89.
[11]HOBZA P,HAVLAS Z.Improper,blue-shifting hydrogen bond[J].Theor Chem Acc,2002,108:325-334.
[12]周杰,王一波.精确计算氢键结合能的相关一致性基函数两点外推法[J].计算机与应用化学,2011,28(9):1112-1116.
[13]张愚,王一波,孙泽民,等.高级量子化学从头计算法研究N2和H2O分子间相互作用[J].高等学校化学学报,2000,21(1):99-104.
[14]吴功兵,于健康,吴迪,等.COCl2…NH3和COCl2…H2S体系的理论研究[J].高等学校化学学报,2006,27(11):2171-2174.
[15]FRISCH M J,TRUCKS G W,SCHLEGEL H B,et al.GAUSSIAN 03[CP].Gaussian,Inc.,Pittsburgh,2003.
[16]许惠英,王维.镁卟啉与氮、氧杂环化合物的相互作用[J].物理化学学报,2011,27(11):2565-2570.
[17]BIEGLER-KÖNIG F,SCHÖNBOHM J,BAYLES D.AIM2000-A program to analyze and visualize atoms in molecules[J].J Comp Chem,2001,22:545-559.The software AIM2000was downloaded at:http://gauss.fh-bielefeld.de/aim2000.
[18]POPELIER P L A.Characterization of a dihydrogen bond on the basis of the electron density[J].J Phys Chem A,1998,102:1873-1878.
[19]KEVIN A F.Role of electrostatic potential in the in silico prediction of molecular bioactivation and mutagenesis[J].J Am Chem Soc,2013,10:1171-1182.
[20]张锁秦,封继康,任爱民,等.取代吡啶系列分子的光谱和二阶非线性光学性质的理论研究[J].高等学校化学学报,1999,20(11):1754-1758.
[21]阚玉和,朱玉兰,侯丽梅,等.含氯不对称配体8-羟基喹啉铝配合物电子和光谱性质的TDDFT研究[J].化学学报,2005,63(14):1263-1268.
[22]卢天.Multiwfn:多功能波函数分析程序[C].//中国化学会第28届学术年会论文集,2012,1.
[23]许惠英,王维,邹建卫,等.PH2X与五元杂环体系磷键相互作用的理论研究[J].化学学报,2013,71(8):1175-1182.
Theoretical studies on hydrogen bonding action mechanism and electronic properties of the base in nucleic acid
XIAO Yi*,FANG Yi,LIU Yuzhen,MA Feiyan,JI Lifei
(CollegeofChemistryandChemicalEngineering,SouthwestUniversity,Chongqing400715,China)
The interactions between pyrimidine and purine were investigated with CCSD/aug-ccpVDZ quantum chemical method,the interaction energies were corrected while basis function equilibrium correction method was adopted to eliminate the overlapping error of the base groups,and the geometric structure was optimized.Besides,the second-order stabilization energies and natural bond orbitals were analyzed with NBO program attached in Gaussian 03 program package.In the meantime,for further exploration of the interactions between pyrimidine and purine,reduced density gradient color-filled iso-surface map was adopted for graphical analyses of pyrimidine-purine system and visualizing the positions and strengths of hydrogen bonding as well as revealing the nature of hydrogen bonding interactions.Results indicate that the interactions of pyrimidine-purine system belong to closed-shell electrostatic interactions.E-lectron density transition matrix analysis shows that the excitation area mainly concentrates on N and O atoms and involves a very large breadth of space.Moreover,the first excited state mainly involves the frontier molecule orbitals,belonging toσ→π*or n→π*type transition.
nucleic acid;base group;hydrogen bonding action mechanism;electronic properties;theoretical study
O 641
A
1008-1011(2014)02-0187-08
2013-11-18.
国家自然科学基金(20873013).
肖 祎(1980-),男,研究员,主要从事量子化学研究.*
,E-mail:xiaoyiswu@sina.com.
猜你喜欢
广州化工(2022年19期)2022-11-09
广州化工(2022年18期)2022-10-22
国防科技大学学报(2020年6期)2020-12-07
教学考试(高考生物)(2020年6期)2020-11-23
硅酸盐通报(2020年1期)2020-02-25
食品与生物技术学报(2020年8期)2020-01-06
测绘通报(2019年11期)2019-12-03
赤峰学院学报·自然科学版(2019年5期)2019-09-10
科学24小时(2019年5期)2019-06-11
发明与创新(2019年9期)2019-03-26
