
Amyloid precursor-like protein 2 C-terminal fragments upregulate S100A9 gene and protein expression in BV2 cells
2014-04-07 02:42GuangzheLiHuiChenLinChengRongjieZhaoJunchangZhaoYanjiXu
中国神经再生研究(英文版) 2014年21期
Guangzhe Li, Hui Chen, Lin Cheng, Rongjie Zhao, Junchang Zhao, Yanji Xu
1 Department of Psychology, Yanbian Brain Hospital, Yanji, Jilin Province, China
2 Department of Preventive Medicine, Medical College, Yanbian University, Yanji, Jilin Province, China
3 Department of Pharmacology, Mudanjiang Medical University, Mudanjiang, Heilongjiang, China
Amyloid precursor-like protein 2 C-terminal fragments upregulate S100A9 gene and protein expression in BV2 cells
Guangzhe Li1, Hui Chen2, Lin Cheng2, Rongjie Zhao3, Junchang Zhao3, Yanji Xu2
1 Department of Psychology, Yanbian Brain Hospital, Yanji, Jilin Province, China
2 Department of Preventive Medicine, Medical College, Yanbian University, Yanji, Jilin Province, China
3 Department of Pharmacology, Mudanjiang Medical University, Mudanjiang, Heilongjiang, China
The murine microglial cell line BV2 has neuroprotective effects, but is toxic to neurons by secreting in fl ammatory cytokines, and is an important target in the treatment of nerve in fl ammation and neurodegenerative diseases. In the present study, we observed the effects of transfecting three amyloid precursor-like protein 2 (APLP2) C-terminal fragments (CTFs; C57, C50 and C31) in the pEGFP-N1 vector on S100A9 expression in BV2 cells. Reverse transcription-PCR, western blot assay and immunocytochemistry revealed that S100A9 protein and mRNA expression was greater in BV2 cells after CTF transfection than after mock transfection with an empty vector. Furthermore, transfection of full-length APLP2-751 resulted in low levels of S100A9 protein expression. Our results show that APLP2-CTFs upregulate S100A9 protein and mRNA expression in BV2 cells, and identify a novel pathway involved in neuronal injury and apoptosis, and repair and protection in Alzheimer’s disease.
nerve regeneration; neurodegeneration; Alzheimer’s disease; APLP2; S100A9; C-terminal fragments; amyloid precursor protein; BV2 cells; γ-secretase; NSFC grant; neural regeneration
Funding:This work was supported by the Natural Science Foundation of Technology Gallery in Jilin Province of China, No. 2011-15237; and the National Natural Science Foundation of China, No. 81160159.
Li GZ, Chen H, Cheng L, Zhao RJ, Zhao JC, Xu YJ. Amyloid precursor-like protein 2 C-terminal fragments upregulate S100A9 gene and protein expression in BV2 cells. Neural Regen Res. 2014;9(21):1923-1928.
Introduction
Various hypotheses exist for the pathogenesis of Alzheimer’s disease, including cholinergic system dysfunction, aberrant inflammatory or immune responses, gene mutations, oxidative stress, excitotoxicity, apoptosis, and defective metabolism of amyloid precursor protein (APP) (Selkoe et al., 2001; Chang et al., 2005; Walker et al., 2006; Ma et al., 2012; Dong and Chai, 2013). Among these, the APP hypothesis has received much attention (Liu et al., 2002; Scheinfeld et al., 2002; Chang et al., 2006). C-terminal fragments of APP may play a contributing role in the pathogenesis of Alzheimer’s disease (Cao and Sudhof, 2001; Kimberly et al., 2001; Minopoli et al., 2001). Amyloid precursor-like protein (APLP) 2 C-terminal fragments (CTFs) were found to translocate to the nucleus and form a ternary complex with the nuclear adaptor protein Fe65 and the transcription factor CP2/LSF/ LBP1, subsequently inducing glycogen synthase kinase-3β expression, tau phosphorylation and apoptosis (Ferrer et al., 2002; Liu et al., 2002; Kim et al., 2003; Xu et al., 2007). APP is a highly conserved gene, and APP, APLP1 and APLP2 have been identi fi ed in mammals. APP contains an amyloid-beta region, but none of its homologues contain this domain, although their amino acid sequence, domain structure and protein organization are similar to APP (Gu et al., 2001; Galvan et al., 2002; Scheinfeld et al., 2002).
APLP2 matures through the same unusual secretory/cleavage pathway as APP, and sheds its extracellular domain in multiple cell culture systems (Zimmer et al., 2005). APLP2 is cleaved by γ-secretase, ε-secretase and caspases to generate the CTFs C57, C50 and C31, all of which contribute to the pathology of Alzheimer’s disease (Gu et al., 2001; Xu et al., 2001; Galvan et al., 2002).
S100A9 is an inflammation-associated calcium-binding protein belonging to the S100 family (Abe et al., 1999; Gebhardt al., 2006; Zhang et al., 2012). Neurological diseases such as cerebral ischemia, traumatic brain injury (Postler et al., 1977) and Alzheimer’s disease (Shepherd et al., 2006; Chang et al., 2012; Wang et al., 2014) are associated with altered expression of S100 family members (Zimmer et al., 2005). S100A9 expression was recently found to be increased within neuritic plaques and reactive glia, and was proposed to participate in the inflammation of Alzheimer’s disease (Shepherd et al., 2006; Zhang et al., 2012; Kim et al., 2014). However, the detailed molecular mechanisms of these patho-logical events remain unknown (Kummer et al., 2006; Lee et al., 2012). TheS100A9gene is signi fi cantly upregulated in the brains of animal models of Alzheimer’s disease, namely Tg2576 and CT-Tg mice, as well as in patients with Alzheimer’s disease (Tae-Young et al., 2010; Chang et al., 2012; Kim et al., 2014). Murine BV2 microglial cells are central nervous system immune cells that protect brain tissue function by phagocytosing neuronal pathogens and harmful particles. However, reactive microglia also release in fl ammatory factors such as S100A9, and are an important target in the treatment of neuroin fl ammation and neurodegenerative diseases. In the present study, we used BV2 cells to investigate the effects of APLP2-CTFs on the expression of the S100A9 gene, to elucidate this pathogenic pathway in Alzheimer’s disease.
Materials and Methods
Materials
APLP2-CTFs and the BV2 cell line were kindly provided by Professor Yoo-hun Suh, Department of Pharmacology, College of Medicine, Seoul National University, South Korea.
Plasmid extract and puri fi cation
Escherichia coli (DH5a; BS-3236, Biomedal, Beijing, China) with recombinant plasmid pEGFP-N1-APLP2-CTFs (C57, C50, C31) and EGFP-N1 empty vector (Clontech, Mountain View, CA, USA) were cultured in medium containing 30 μg/mL kanamycin (Kan+). A single colony of each APLP2-CTF was selected into 10 mL Luria-Bertani liquid medium with Kan+, and mixed overnight on a rotary shaker at 230 r/min and 37°C. The colonies were then transferred to 200 mL of Luria-Bertani liquid medium to grow. The plasmids were extracted using the Plasmid Maxi Kit (12163; QIAGEN, Venlo, Netherlands) and the concentration and purity were measured in an ultraviolet spectrophotometer (CKX41; Olympus, Tokyo, Japan).
BV2 cell culture
Murine microglial BV2 cells (Department of Pharmacology, Seoul National University, South Korea) were cultured in Dulbecco’s modi fi ed Eagle’s medium (DMEM; Life Technologies Inc., Grand Island, NY, USA) in 6-well plates, supplemented with 5% fetal bovine serum (Corning, Steuben Country, NY, USA) and penicillin/streptomycin (100 U/mL/100 μg/mL) at 37°C and 5% CO2for 3 days (Munsie et al., 2011; Lorena et al., 2006).
Plasmid transfection
At this point, the culture was divided into four groups: a mock transfection group (pEGFP-N1 empty vector); an APLP2-C57 transfection group; an APLP2-C50 transfection group; and an APLP2-CT31 transfection group. Each group was transfected with 5 μg plasmid for 48 hours and observed 24 hours post-transfection. BV2 cell cultures were maintained in 24-well plates. In addition, to inhibit CTF generation, BV2 cells were transfected with full-length APLP2-751 and incubated with an inhibitor of γ-secretase, N-[N-(3,5-difluorophenacetyl)-l-alanyl]-S-phenylglycine t-butyl ester (DAPT, 2 μmol/L; Sigma-Aldrich, St Louis, MO, USA). All transfection procedures were carried out using 5 μL Lipofectamine™ 2000 (Invitrogen, Carlsbad, CA, USA) in 1 mL DMEM, according to the manufacturer’s instructions. Cells were observed under a fl uorescence microscope (Olympus, Tokyo, USA).
Western blot assay
Samples were harvested in radioimmunoprecipitation assay buffer (1% Triton X-100, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 0.15 mol/L NaCl, and 0.05 mol/L Tris-HCl; pH 7.2) with protease inhibitors (Complete mini; Roche, Indianapolis, IN, USA) at 4°C. The harvested proteins from cell lysates were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to a polyvinylidene difluoride membrane. The membrane was blocked in Tris-buffered saline containing 4% non-fat milk powder (Lise et al., 2012) for 1 hour at room temperature. The proteins were visualized using activated rabbit anti-S100A9 monoclonal antibody overnight (1:1,000; Sino Biological Inc.) followed by horseradish peroxidase-conjugated goat anti-rabbit IgG (H + L) for 1 hour at room temperature (1:800; Beyotime Institute of Biotechnology, Haimen, Jiangsu Province, China). The membrane was scanned in an ultraviolet spectrophotometer (Olympus) and the gray values of the target protein bands were compared against those of β-tubulin (mouse, 1:1,000; Sino Biological Inc.; 1 hour at room temperature) using Quantity One software (Bio Rad, Hercules, CA, USA).
Reverse transcription (RT)-PCR
Trizol reagent (Invitrogen) was used to isolate 2 mg total RNA for cDNA synthesis, which was carried out using Accu-Power RT PreMix (Bioneer, Korea). We measured the concentration and purity of RNA by ultraviolet spectrophotometry (CKX41; Olympus). The abundance of transcripts in cDNA samples was measured by RT-PCR (Waters Company, Changchun, Jilin Province, China). The primer sequences are as follows:

PrimerGene sequence Product size (bp) S100A9 Forward 5′- CAG CAT AAC CAC CAT CAT CG-3′362 Reverse 5′-GTC CTG GTT TGT GTC CAG GT-3′ ; β-Actin Forward 5′-CCA GAT CAT GTT TGA GAC CT-3′206 Reverse 5′-GTT GCC AAT AGT GAT GAC CT-3′
The PCR reactions were subjected to 40 amplification cycles of 95°C for 1 minute, 58°C for 1 minute, and 72°C for 1 minute. PCR products were analyzed on 1.5% agarose gel, and the gels were visualized by staining with ethidium bromide. All absorbance values were normalized to β-actin.

Figure 1 Effects of transfection of BV2 cells with APLP2-CTFs (C57, C50, C31) on S100A9 protein expression (western blot assay).

Figure 3 Effects of transfection with APLP2-CTFs (C57, C50, C31) onS100A9mRNA expression in BV2 cells (reverse transcription-PCR).

Figure 4 Effects of transfection with APLP2-751 (full-length) on S100A9 protein expression in BV2 cells.
Immunocytochemistry
BV2 cells grown on a glass coverslip were transfected with APLP2-CTFs for 48 hours. Cells were fixed in 4% paraformaldehyde for 1 hour at 4°C and then for 30 minutes at room temperature. After quenching in 50 mmol/L NH4Cl in phosphate-buffered saline (PBS) for 10 minutes, the cells were washed in PBS and permeabilized for 30 minutes at room temperature in permeabilization buffer (PBS containing 0.1% Triton X-100 and 1 mg/mL bovine serum albumin). The following steps were performed at room temperature in permeabilization buffer. Cells were incubated with rabbit anti-S100A9 antibody (1:1,000; Sino Biological Inc.) for 1 hour at room temperature, washed three times, and incubated with Alexa Fluor 555-labeled goat anti-rabbit IgG (H+L; Institute of Biotechnology; 1:800 dilution) for 1 hour at room temperature. After three washes in permeabilization buffer and one wash in PBS, the cells were mounted on microscope slides in mounting medium (DAKO, Copenhagen, Denmark) containing DAPI (1:50; Invitrogen) for nuclear staining. Cells were observed under a laser scanning confocal microscope (Leica TCS SP5, Leica Microsystems, provided by the Experimental Center of Preventive Medicine, Medical College, Yanbian University, China). Data were analyzed using STED deconvolution software (LAS AF; Leica, Germany).
Statistical analysis
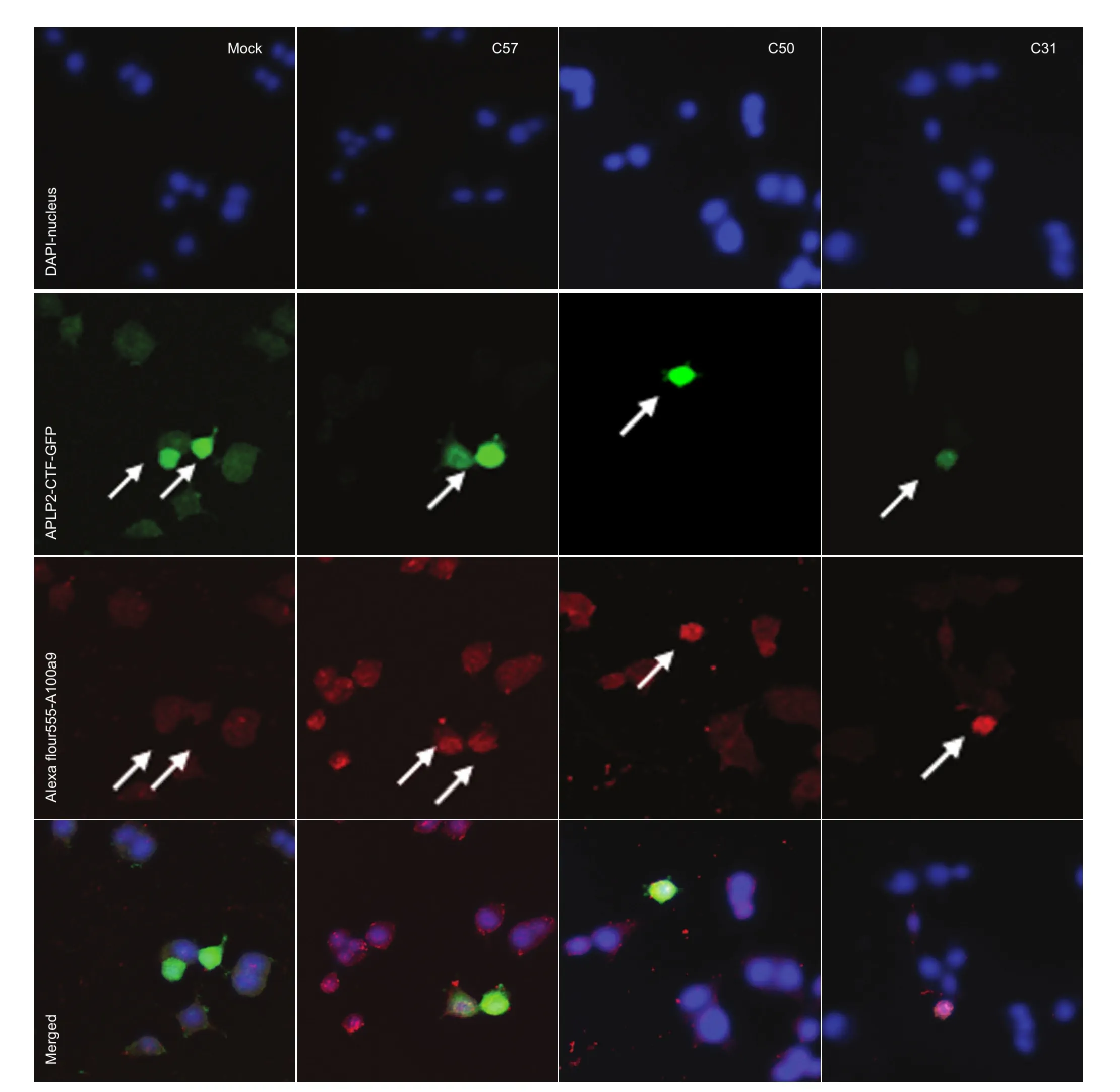
Figure 2 Effects of transfection with APLP2-CTFs (C57, C50, C31) on S100A9 protein expression in BV2 cells (immunocytochemical staining; laser scanning confocal microscope, × 100).
All data were expressed as the mean ± SD of at least three independent experiments. Pairedt-tests, or one-way analysis of variance followed by Dunnett’spost-hoct-test, were performed using SPSS 13.0 software (SPSS, Chicago, IL, USA) to study the relationship between the different variables. Values ofP< 0.05 were considered statistically signi fi cant.
Results
APLP2-CTFs induced upregulation of S100A9 protein expression
BV2 cells were transfected with APLP2-CTFs for 48 hours. Western blot assay revealed that expression of S100A9 was signi fi cantly greater in all three groups of APLP2-CTF-transfected cells than in the mock transfected group (P< 0.05; Figure 1).
Immunocytochemical staining con fi rmed that S100A9 was induced by APLP2-CTFs (Figure 2).
APLP2-CTFs induced upregulation of S100A9 mRNA
RT-PCR showed that S100A9 expression was significantly greater in all three APLP2-CTF-transfected groups than inthe mock transfected group (P< 0.05; Figure 3).
DAPT treatment induced downregulation of S100A9 protein levels
To determine whether endogenously generated APLP2-CTFs affect S100A9 expression, we investigated the effect of DAPT, an inhibitor of γ-secretase, on S100A9 levels after transfection with full-length APLP2-751. We found that S100A9 expression was lower in the DAPT treatment group than in the vehicle-treated control group (P< 0.05), indicating that the generation of CTFs is critical for S100A9 protein expression (Figure 4).
Discussion
Alzheimer’s disease is characterized by age-dependent formation of amyloid-beta-containing senile plaques and intracellular neuro fi brillary tangles (Han et al., 2013; Wang et al., 2013). Induction of theS100A9gene occurs in the brains of patients with Alzheimer’s disease and in animal models of the disease. S100A9 expression is increased in senile plaques and active gliocytes, suggesting that S100A9 is involved in the pathogenesis of Alzheimer’s disease (Zimmer et al., 2005; Shepherd et al., 2006; van Lent et al., 2008). In the present study, we found that transfecting BV2 cells with APLP2-CTFs increased S100A9 protein and mRNA expression.
Previous studies showed thatS100A9gene silencing significantly reduces Alzheimer’s disease pathology, including the number of plaques, and improves the learning ability of Tg2576 mice. Also, treatment with amyloid-beta (1-42) or C105 and transfection with APP-C50 or C99 can elevateS100A9mRNA expression (Tae-Young et al., 2010). Our results demonstrate that APLP2-CTF has similar toxic effects to those of APP-CTF and amyloid beta on the in fl ammatory responses of glial cells.
The amyloid precursor protein cleaved by β-secretase and γ-secretase generates amyloid beta and CTFs of different sizes (Xing et al., 2012; Liu et al., 2013). APP-CFTs play a contributing role in Alzheimer’s disease pathogenesis (Cao and Sudhof, 2001; Kimberly et al., 2001; Minopoli et al., 2001; Kim et al., 2003). APLP2-CTFs translocate to the nucleus and form a ternary complex with Fe65 and CP2/LSF/LBP1, and induce the expression of glycogen synthase kinase-3β, tau phosphorylation and apoptosis (Xu et al., 2007).
We also examined changes in S100A9 levels in BV2 cells expressing full-length APLP2-751 in the presence or absence of the γ-secretase inhibitor, DAPT, to investigate the effects of endogenously generated APLP2-CTFs on S100A9 expression. In the DAPT-treated group, S100A9 levels were not signi fi cantly greater than those in the control group, suggesting that generation of APLP2-CTFs is essential for the induction of S100A9 gene expression in BV2 cells.
The results of the present study indicate that APLP2-CTF co-contributes to the pathology of Alzheimer’s disease with APP-CTF and amyloid beta. The underlying molecular mechanisms need to be further investigated.
Acknowledgments:APLP2-CTFs and BV2 cell line kindlyare provided by Professor Suh YH from Department Pharmacology, College of Medicine, Seoul National University South Korea.
Author contributions:Li GZ and Xu YJ participated in conception and design of the study, and analysis and interpretation of the data. Li GZ, Chen H and Cheng L were in charge of data collection. Chen H, Li GZ, Zhao RJ and Xu YJ wrote the manuscript or provided critical revision of the manuscript for intellectual content. Zhao JC participated in statistical expertise. All authors approved the final version of the paper.
Con fl icts of interest:None declared.
Abe M, Umehara F, Kubota R, Moritoyo T, Izumo S, Osame M (1999) Activation of macrophages microglia witn the calcium-binding protein MRP14 and MRP8 is related to the lesional activities in the spinal cord of HTLV-I associated myelopathy. N J Neurol 246:358-364.
Cao X, Sudhof TC (2001) A transcriptionally active complex of APP with Fe65 and histone acetyltransferase Tip60. Science 293:115-120.
Chang KA, Kim HS, Ha TY, Ha JW, Shin KY, Jeong YH, Lee JP, Park CH, Kim S, Baik TK, Suh YH (2006) Phosphorylation of amyloid precursor protein (APP) at Thr668 regulates the nuclear translocation of the APP intracellular domain and induces neurodegeneration. Mol Cell Biol 26:4327-4338.
Chang KA, Kim HJ, Suh YH (2012) The role of S100A9 in the pathogenesis of Alzheimer’s disease: the therapeutic effects of S100A9 knockdown or knockout. Neurodegener Dis 2012;10:27-29.
Chang KA, Suh YH (2005) Pathophysiological roles of amyloidogenic carboxyterminal fragments of the beta-amyloid precursor protein in Alzheimer’s disease. J Pharmacol Sci 97:461-471.
Dong XH, Chai XQ (2013) Alzheimer’s disease transgenic animal models: How to get more similar pathological characteristics? Zhongguo Zuzhi Gongcheng Yanjiu 17:8075-8082.
Ferrer I, Barrachina M, Puig B (2002) Glycogen synthase kinase-3 is associated with neuronal and glial hyperphosphorylated tau deposits in Alzheimer’s disease, Pick’s disease, progressive supranuclear palsy and corticobasal degeneration. Acta Neuropathol (Berl) 104:583-591.
Galvan V, Chen S, Lu D, Logvinova A, Goldsmith P, Koo EH, Bredesen DE (2002) Caspase cleavage of members of the amyloid precursor family of proteins. J Neurochem 82:283-294.
Gebhardt C, Nemeth J, Angel P, Hess J (2006) S100A8 and S100A9 in in fl ammation and cancer. Biochem Pharmacol 72:1622-1631.
Gu Y, Misonou H, Sato T, Dohmae N, Takio K, Ihara Y (2001) Distinct intramembrane cleavage of the beta-amyloid precursor protein family resembling gamma-secretase-like cleavage of Notch. J Biol Chem 276:35235-35238.
Ha TY, Chang KA, Kim Ja, Kim HS, Kim SH, Chong YH, Suh YH (2010) S100A9 knockdown decreases the memory lmpairment and the neuropathology in Tg2576 mice, AD animal model. PLoS One 5:1-11.
Han HY, Zhang YC, Ji SQ, Liang QM, Zhu SQ, Xue Z (2013) Integrin beta 1 inhibits long-term potentiation induced by amyloid beta-protein. Zhongguo Zuzhi Gongcheng Yanjiu 17:1959-1964.
Kim HJ, Chang KA, Ha TY, Kim J, Ha S, Shin KY, Moon C, Nacken W, Kim HS, Suh YH (2014) S100A9 knockout decreases the memory impairment and neuropathology in crossbreed mice of Tg2576 and S100A9 knockout mice model. PLoS One. 9:e88924.
Kim HS, Kim EM, Lee JP, Park CH, Kim SH, Seo JH, Chang KA, Yu EA, Jeong SJ, Chong YH, Suh YH (2003) C-terminal fragments of amyloid precursor protein exert neurotoxicity by inducing glycogen synthase kinase-3beta expression. FASEB J 17:1951-1953.
Kimberly WT, Zheng JB, Guenette SY, Selkoe DJ (2001) The intracellular domain of the beta-amyloid precursor protein is stabilized by Fe65 and translocates to the nucleus in a notch-like manner. J Biol Chem 276:40288-40292.
Kummer MP, Vogl T, Axt D, Griep A, Vieira-Saecker A, Jessen F, Gelpi E, Roth J, Heneka MT (2012) Mrp14 de fi ciency ameliorates amyloid β burden by increasing microglial phagocytosis and modulation of amyloid precursor protein processing. J Neurosci 32:17824-17829.
Lee EO, Yang JH, Chang KA, Suh YH, Chong YH (2013) Amyloid-β peptide-induced extracellular S100A9 depletion is associated with decrease of antimicrobial peptide activity in human THP-1 monocytes. J Neuroin fl ammation 10:68.
Lise NM, Carly RD, Ray T (2012) Co fi lin nuclear-cytoplasmic shuttling affects co fi lin-actin rod formation during stress. J Cell Sci 125:3977-3988.
Liu F, Lqbal K, Grundke-lqbal I, Gong CX (2002) Involvement of aberrant glycosylation in phosphorylation of tau by cdk5 and GSK-3beta. FEBS Lett 530:209-214.
Liu R, Hou H, Yi X, Wu S, Zeng H (2013) Divalent cation tolerance protein binds to β-secretase and inhibits the processing of amyloid precursor protein. Neural Regen Res 8:991-999.
Lorena H, Pablo H, Soledad DO, Gabriela K, Francisco SV, Frank LF, Matthias S, Jose DO, Jorge B, Alfredo C, Alfredo L (2006) Phosphorylation of actin-depolymerizing factor/co fi lin by lim-kinase mediates amyloid-induced degeneration: a potential mechanism of neuronal dystrophy in Alzheimer’s disease. J Neurosci 26:6533-6542.
Ma HP, Lü XR, Shi HY, Shi ZX, Li XC, Li Z (2012) Effect of nerve growth factor pretreatment on phosphorylated Tau protein expression in hippocampal neurons in Alzheimer’s disease rats. Zhongguo Zuzhi Gongcheng Yanjiu 16:8641-8646.
Minopoli G, de Candia P, Bonetti A, Faraonio R, Zambrano N, Russo T (2001) The beta-amyloid precursor protein functions as a cytosolic anchoring site that prevents Fe65 nuclear translocation. J Biol Chem 276:6545-6550.
Munsie L, Caron N, Atwal RS, Marsden I, Wild EJ, Bamburg JR, Tabriz SJ, Truant R (2011) Mutant huntingtin causes defective actin remodeling during stress: defining a new role for transglutaminase 2 in neurodegenerative disease. Hum Mol Genet 20:1937-1951.
Postler E, Lehr A, Schluesener H, Meyermann R (1977) Expression of the S-100 protein MRP-8 and -14 in ischemic brain lesions. Glia 19:27-34.
Selkoe KJ (2001) Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev 81:41-766.
Scheinfeld MH, Ghersi E, Laky K, Fowlkes BJ, D’Adamio L (2002) Processing of beta -amyloid precursor-like protein-1 and -2 by gamma-secretase regulates transcription. J Biol Chem 277:44195-44201.
Shepherd CE, Goyette J, Utter V, Rahimi F, Yang Z, Geczy CL, Halliday GM (2006) Inflammatory S100A9 and S100a12 proteins in Alzheime’s disease. Neurobiol Aging 27:1554-1563.
van Lent PL1, Grevers L, Blom AB, Sloetjes A, Mort JS, Vogl T, Nacken W, van den Berg WB, Roth J (2008) Myeloidrelated proteins S100A8/ S100A9 regulate joint in fl ammation and cartilage. destruction during antigen-induced arthritis. Ann Rheum Dis 67:1750-1758.
Walker DG, Link J, Lue LF, Dalsing-Hernandez JE, Boyes BE (2006) Gene expression changes by amyloid beta peptide-stimulated human postmortem brain microglia identify activation of multiple in fl ammatory processes. J Leukoc Biol 79:596-610.
Wang C, Klechikov AG, Gharibyan AL, Wärmländer SK, Jarvet J, Zhao L, Jia X, Shankar SK, Olofsson A, Brännström T, Mu Y, Gräslund A, Morozova-Roche LA (2014) The role of pro-in fl ammatory S100A9 in Alzheimer’s disease amyloid-neuroinflammatory cascade. Acta Neuropathol 127:507-522.
Wang W, Zhang HT, Wang SH, Zhang YB (2013) Correlation of Alzheimer’s disease with Wnt signaling pathway and neural stem cells. Zhongguo Zuzhi Gongcheng Yanjiu 17:3566-3572.
Xing SL, Chen C, Shen DZ (2012) Signal pathway of amyloid precursor protein in neuronal cell injury induced by amyloid-beta peptide 1-42. Zhongguo Zuzhi Gongcheng Yanjiu 16:2797-2800.
Xu KP, Zoukhri D, Zieske JD, Dartt DA, Sergheraert C, Loing E, Yu FS (2001) A role for MAP kinase in regulating ectodomain shedding of APLP2 in corneal epithelial cells. Am J Physiol Cell Physiol 281:C603-614.
Xu YJ, Kim HS, Joo Y, Choi Y, Chang KA, Park CH, Shin KY, Kim S, Cheon YH, Baik TK, Kim JH, Suh YH (2007) Intracellular domains of amyloid precursor-like protein2 interact with CP2 transcription factor in the nucleus and induce glycogen synthase kinase-3β expression. Cell Death Differ 14:79-91.
Zimmer DB, Chaplin J, Baldwin A, Rast M (2005) S100-mediated signal transduction in the nervous system and neurological diseases. Cell Mol Biol (Noisy-le-grand) 51:201-214.
Zhang C, Liu Y, Gilthorpe J, van der Maarel JR (2012) MRP14 (S100A9) protein interacts with Alzheimer beta-amyloid peptide and induces its fi brillization. PLoS One 7:e32953.
Copyedited by Slone-Murphy J, Norman C, Wang J, Qiu Y, Li CH, Song LP, Zhao M
Yanji Xu, Ph.D., Department of Preventive Medicine, Medical College, Yanbian University, Yanji 133002, Jilin Province, China, xuyanji@ybu.edu.cn.
10.4103/1673-5374.145362
http://www.nrronline.org/
Accepted: 2014-09-06

- 中国神经再生研究(英文版)的其它文章
- Reversible lesions in the brain parenchyma in Wilson’s disease con fi rmed by magnetic resonance imaging: earlier administration of chelating therapy can reduce the damage to the brain
- Effects of diazepam on glutamatergic synaptic transmission in the hippocampal CA1 area of rats with traumatic brain injury
- The occurrence of diffuse axonal injury in the brain: associated with the accumulation and clearance of myelin debris
- Adult neurogenesis in the four-striped mice (Rhabdomys pumilio)
- Recovery of cerebellar peduncle injury in a patient with a cerebellar tumor: validation by diffusion tensor tractography
- Hot spots and future directions of research on the neuroprotective effects of nimodipine