
模拟环境条件下δ-MnO2氧化As(III)的搅拌流动动力学特征
2014-05-23 08:24夏平平谭文峰邱国红冯雄汉华中农业大学资源与环境学院农业部长江中下游耕地保育重点实验室湖北武汉430070
中国环境科学 2014年4期
王 鹍,夏平平,刘 凡,谭文峰,邱国红,冯雄汉 (华中农业大学资源与环境学院,农业部长江中下游耕地保育重点实验室,湖北 武汉 430070)
模拟环境条件下δ-MnO2氧化As(III)的搅拌流动动力学特征
王 鹍,夏平平,刘 凡,谭文峰,邱国红,冯雄汉*(华中农业大学资源与环境学院,农业部长江中下游耕地保育重点实验室,湖北 武汉 430070)
采用搅拌流动法研究了酸性水钠锰矿及水羟锰矿2种δ-MnO2矿物氧化As(Ⅲ)的动力学过程,构建了可用于多相体系的搅拌-流动氧化还原反应动力学模型.经过As吸附量的校正后,该模型对酸性水钠锰矿及水羟锰矿氧化As(III)动力学数据拟合度分别为0.980和0.951,模型拟合得到pH 7时2种矿物单位比表面上氧化As(III)的初始反应速率常数k分别为0.131,0.014min-1·m-2.相比而言,该速率常数明显高于批量法得到的表观速率常数kobs,0.021,0.001min-1·m-2更接近真实的化学动力学参数.搅拌流动法与批量法得到的不同矿物的速率常数大小趋势一致,即尽管酸性水钠锰矿对 As(III)的氧化率低于水羟锰矿,单位比表面上酸性水钠锰矿氧化 As(III)初始反应速率却远高于水羟锰矿.反应过程分析表明,反应初始阶段,As(III)的吸附为主要限速步骤;而随着反应的进行,矿物表面反应位点逐渐钝化或减少,反应位点数量成为限速步骤.
As(III);δ-MnO2;搅拌流动法;动力学;速率常数;反应位点
近年来,采矿、制革和农业等行业生产过程中产生了大量含砷废水、废渣,对土壤和水体环境产生了严重污染.对于人体健康而言,As(III)的毒性和移动性远远大于 As(V)[1-4].δ-MnO2是土壤、沉积物及大洋锰结核中最为常见的氧化锰矿物,其结晶度弱、比表面积高,是某些特定金属As(III)和 Cr(III)的天然氧化剂[4-7].因而它们对土壤中砷的形态转化和化学迁移过程有着重要的调控作用.
在非均相多介质环境体系中,As(III)的反应动力学可以成为影响和决定其环境过程的主导因素.目前报道主要是采用批量法研究静态体系中整个反应过程的表观动力学特性,难以揭示表面反应本身的动力学过程和机制.与表观动力学相对应的是化学动力学,是研究不受分子运移、副反应等因素影响的理想条件下所发生的化学反应的动力学过程[8].化学动力学可以了解和预测单一化学反应速率、限速步骤和动力学常数,借以揭示其反应机理.常见的土壤化学动力学研究方法有批量法、流动法和驰豫法.弛豫法常适用于毫秒、微秒数量级反应时间的离子交换等快速反应过程.Tournassatat等[9]通过批量法研究水钠锰矿氧化 As(III)的过程,但该法未考虑反应生成物表面沉淀和逆反应等的影响.Ginder-Vogel[10]运用快速动力学方法原位得到了 1~60s As(III)氧化动力学参数,但仍建立在批量法基础之上.Zhu等[11]通过密度函数理论的方法来模拟层状锰氧化物吸附氧化 As(III)的反应机制,然而缺乏实验数据支撑.搅拌流动体系能较好地模拟实际土壤开放的流动状态,且大大减少因产物积累引起的逆反应和副反应,因此更接近化学动力学过程[12].在土壤和环境化学研究中常通过搅拌流动法模拟田间的离子交换和吸附/解吸动力学[13-15],近年来也有搅拌流动法研究锰矿物氧化还原体系的报道,如一系列反应阶段水羟锰矿氧化 As(III)反应历程的研究等[16-18].但对于搅拌流动法氧化反应动力学模型的拟合和定量描述未见报道.
本文采用搅拌-流动法来模拟土壤流动环境,构建可用于多相体系搅拌流动法的氧化动力学模型,推求反应速率常数等动力学参数.同时与批量法得到的表观速率常数进行比较,期望进一步了解δ-MnO2与As(III)的氧化化学动力学过程和反应机制.
1 材料与方法
1.1 矿物合成与表征
酸性水钠锰矿采用浓盐酸还原高锰酸钾方法制备[19].水羟锰矿按 Villalobos[20]的方法制得,样品磨细过60目筛装瓶备用.对样品进行粉晶X衍射分析:合成的矿物按粉末压片,在 Bruker D8Advance X射线衍射仪上进行鉴定分析.测试条件为:CuKα (λ=0.15406nm),管压 40kV,管流40mA,步 进 扫 描 ,步 长 为 0.02°,扫 描 速 度 为10°/min.锰平均氧化度测定采用草酸还原-高锰酸钾返滴定法[21].样品比表面积测定使用全自动比表面和孔径分布分析仪(Quantachrome Autosorb-1,JEDL-6390/LV),称取矿物粉末 0.1 ~0.2g,在110℃脱气3h,采用N2吸附法,测定比表面积.样品形貌通过 JEOL-场发射扫描电子显微镜(JSM6700F, 日本电子)分析.
1.2 δ-MnO2氧化As(III)氧化动力学实验
1.2.1 搅拌流动法实验装置 实验装置由恒温水槽、高效液相泵、磁力搅拌器、反应池和自动溶液收集器组成(图 1).反应开始前,将反应液置于恒温水槽中的容器内,MnO2悬液置于反应池中,反应开始后高效液相恒流泵将反应液泵入反应池(反应池体积为 8.5mL),在磁力搅拌器的作用下与供试样品充分接触后,因内压的作用从反应池的顶部通过0.45μm的微孔滤膜自动流出清液.搅拌流动池的外壁通过循环水,循环水与恒温水槽相连以保证反应过程中恒温.所有反应体系中搅拌速度保持一致.待第一滴溶液流出时打开自动部分收集器,采样时间为1min自动收集溶液.干样矿物固体先形成一定浓度的悬浮液,将上述悬浮液取8.5mL至于反应装置中.

图1 搅拌流动法的实验装置示意Fig.1 The diagram of the stirred-flow experimental setup
1.2.2 搅拌流动法动力学实验 室温(25℃)下,配制酸性水钠锰、水羟锰矿 10g/L浓度的悬浮液, 充分混匀搅拌 12h,以使搅拌速度和矿物的水合作用对实验的影响降至最低.恒定流入液As(III)浓度为 500μmol/L,流速设置为 2mL/min,自动收集反应流出液.测定流出液总As和As(V)和 Mn2+的浓度,得到动力学数据,每次动力学实验重复3次.所有溶液用HNO3或NaOH溶液调节 pH 7±0.03(所有溶液均含背景电解质溶液50mmol/L NaNO3溶液和5mmol/L Mops-Na缓冲溶液).
测定前,用空白支持电解质溶液代替以上锰矿物悬浮液进行试验,并通过稀释曲线模型检验,确保实验条件下反应体系搅拌分散均匀.流出液中As(V)采用砷钼兰比色测定法[22]测定,总 As和总 Mn采用电感耦合等离子体质谱仪(ICP-MS)测定.滤液中总 As和 As(V)二者之差为As(III).
1.2.3 批量法实验 批量法方法易操作,反应条件能较好控制,不需要特殊装置.室温(25℃)下,取0.15g氧化锰矿物在150mL氧化锰矿物在背景电解质溶液中充分悬浮12h,用HNO3或NaOH溶液调节 pH 为 7±0.05,加入 150mL 1mmol/LAs(III)溶液(所有溶液均含背景电解质溶液 50mmol/L NaNO3溶液和5mmol/L Mops-Na缓冲溶液[16]),溶液在加入悬浮液反应之前调节 pH 至初始值7±0.05.控制搅拌速度一定.反应开始后,分别在1,2,3,5,7,10,15,20,30,40,50,60,90,120,180,240,300,360min取样4mL左右.取样后,立即用0.45μm微孔滤膜过滤,滤液保留待测.滤液中总 As和 As(V)、总Mn测定方法同上.
1.3 氧化动力学模型
1.3.1 搅拌流动法氧化动力学模型的构建
1) 稀释曲线理论模型
无氧化锰稀释体系中流出液中稀释曲线动力学是根据质量平衡来建立,即反应池中变化的量 = 流入反应池的量-流出反应池的量.设As(III)的初始浓度为 C0,流出液中 As(III)的浓度为Ct,高效液相泵的流量为F,反应池的体积为V,稀释曲线的理论模型如下:
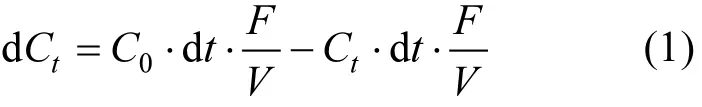
对式(1)积分得:

即得到的稀释曲线的理论方程.
2) 氧化动力学模型构建
搅拌流动法氧化动力学模型也是根据整个体系的质量守恒来建立的,即流入反应池的量 =流出反应池的量 + 反应池中变化的量 + 氧化还原反应的量.反应体系中矿物大大过量,一定时间内其浓度视为常数,即初始氧化速率常数 k保持不变.设 As(III)的初始浓度为 C0,流出液中As(III)的浓度为 Ct,流出液中总砷浓度为 Ct′,高效液相泵的流量为 F,反应池的体积为 V,氧化还原反应的速率常数为 k.忽略 As的吸附,则 As(III)的氧化动力学模型构建如下:
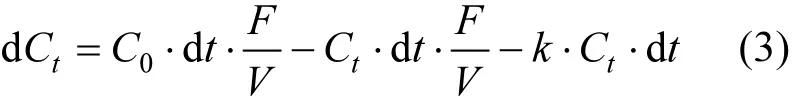
对式(3)积分,则有:

设流出液中As(V)浓度为Ca,则:

将式(4)代入式(5)得:
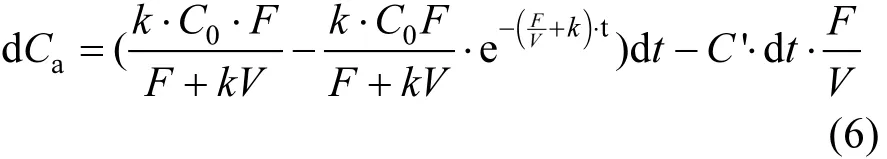
解式微分方程(6)得流出液中 As(V)浓度随时间的变化关系:
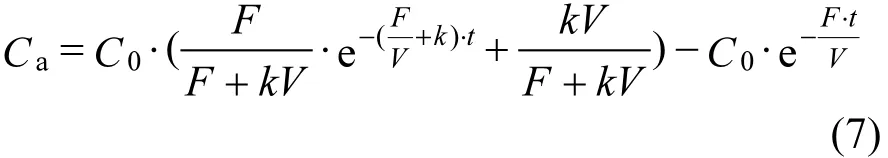
3) 搅拌流动法吸附量
设无氧化锰稀释体系流出液中t时刻As(III)的浓度为 Ct;氧化锰氧化体系中 t时刻流出液中总砷浓度为 Ct′,流出液中 As(V)浓度为 Ca,As吸附量为Qm,则:
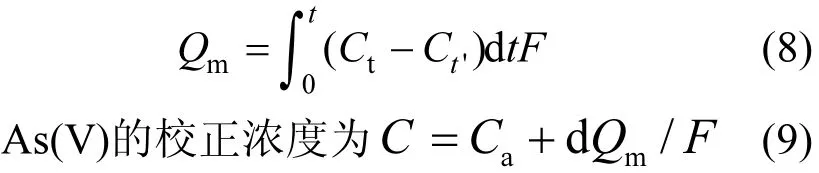
1.3.2 批量法动力学方程
MnO2对 As(III)的氧化反应式可表示为式(10):
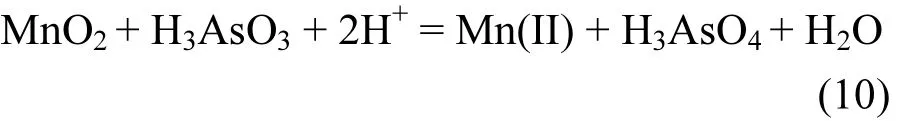
在初始反应阶段,MnO2矿物浓度大大过量,其浓度可视为常数,生成 Mn2+和 H3AsO4对反应速率影响可忽略.缓冲体系中pH值变化较小,H+浓度可视为常数.故初始阶段动力学方程可以简化为如下的准一级动力学方程式:
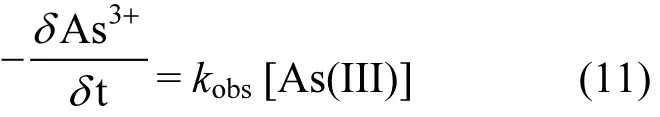
对式(11)积分得:
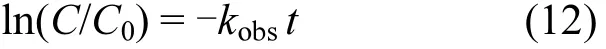
式中:C为 As(III)即时浓度 (mg/L);C0为 As(III)初始浓度(mg/L);t为反应时间 (min).㏑(C/C0)与反应时间t成线性关系,斜率k即为表观速率常数kobs.
2 结果与讨论
2.1 供试氧化锰矿物的性状
酸性水钠锰矿粉晶 X-射线衍射 (XRD)图谱如图2所示,其中0.714,0.357,0.242,0.142nm峰均为水钠锰矿的特征衍射峰(JCPDS 23-1239),没有其他矿物相衍射峰.而水羟锰矿的 X-射线衍射图(XRD)显示有 2个特征衍射峰:0.242,0.142nm,另外2个峰的强度很弱,无其它杂质峰.
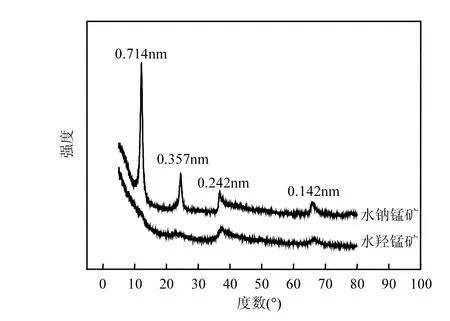
图2 酸性水钠锰矿和水羟锰矿粉晶衍射图谱Fig.2 Powder XRD patterns of birnessite (Bir) and vernadite (Ver)
如图3a所示,水钠锰矿为薄片状晶体团聚在一起,形成花球状集合体,与文献所报道水钠锰矿晶体微观形貌一致[23-26].水钠锰矿结晶程度好,比表面积较小,仅为17.1m2/g.图3b中水羟锰矿呈极细小的弱晶质颗粒状,存在明显的晶粒团聚现象.其结晶程度弱,比表面积大,达到 196.2m2/g.合成的酸性水钠锰矿与水羟锰矿氧化度分别为 3.87和3.84,水钠锰矿氧化度略高于水羟锰矿.

图3 合成氧化锰矿物发射—扫描电镜图(FE—SEM)Fig.3 FE-SEM images of synthesized Mn oxide minerals
2.2 δ-MnO2氧化As(III)氧化动力学实验结果
2.2.1 搅拌流动法实验体系 如图4所示,在流速2mL/min条件下,随着初始溶液不断流入反应池中,在12min时稀释曲线接近初始浓度值,稀释曲线方程拟合R2均在0.997以上,拟合得到初始加入浓度为 496.9μmol/L,表明在本实验的条件下,反应体系搅拌分散均匀,仪器的工作条件非常稳定.也说明后期加入锰矿物反应的流动动力学数据的可靠,仪器等方面的误差以及溶液稀释带来的误差较小.
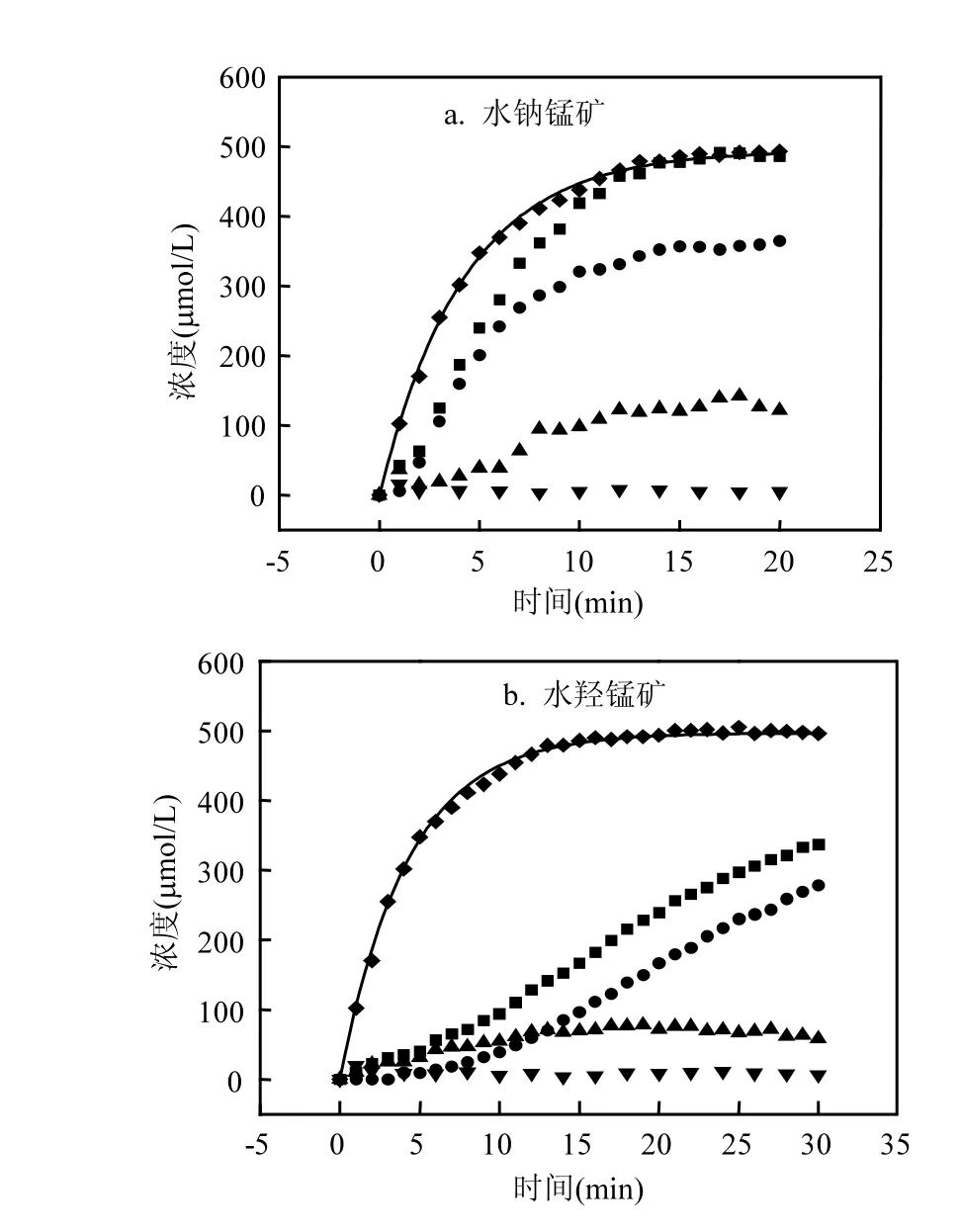
图4 合成氧化锰矿物氧化As(III)的搅拌流动法动力学曲线Fig.4 The kinetic curves of As(III) oxidation by synthesized Mn oxide minerals in stirred flow test
图4a中,水钠锰矿体系中初始阶段As(V)浓度曲线陡峭,氧化速率快,随着反应进行,曲线趋向平坦,As(V)浓度趋于稳定.由图 4b可知,当As(III)与水羟锰矿反应时,有 2个反应阶段,在前5min初始阶段时没有检测到As(V),此时水羟锰矿对生成的 As(V)吸附速率快于解吸速率[16],因而生成的 As(V)基本吸附在矿物表面.尽管流动法在一定程度上可消除反应中产物的影响,然而初始阶段生成的 As(V)量少,而矿物相对大大过量,因此矿物对产物吸附仍然明显,但此时矿物表面位点相对过量,吸附 As(V)对反应影响较小.随后,As(V)浓度逐渐上升趋于稳定.在反应前 3min流出液中都没有检测到 As(III),此时 As(III)或者吸附在矿物表面或者被氧化了,随着反应进行,溶液中 As(III)浓度的逐渐增加趋于稳定.表明此时As(III)的去除(包括吸附氧化)速率一定.而且此时水钠锰矿体系中 As(V)的浓度也趋于恒定,表明氧化速率和 As(V)释放速率趋于平衡,此时浓度高低更能反映MnOx对As的固定和氧化能力.溶液中检测到 Mn2+含量很低,生成的 Mn2+几乎完全吸附,同时也从侧面反映了矿物是过量的.
2.2.2 搅拌流动法动力学方程拟合 δ-MnO2氧化As(III)

图5 合成氧化锰矿物氧化As(III)的动力学模型拟合Fig.5 Fitting of kinetic curves of As(III) oxidation by synthesized Mn oxide minerals with the model
由图5可见,流出液中As(III)的动力学数据拟合较差,主要是由于初始阶段流出液中 As(III)浓度较低,测定误差大.而流出液中 As(V)浓度拟合度也较低,且拟合曲线偏低,这是因为:由稀释体系和氧化体系总 As曲线的差异可知,反应生成的 As(V)部分吸附在矿物表面,这使得流出液中 As(V)的浓度因受到吸附的影响,而偏离忽略As吸附的理论模型,拟合反应动力学参数低于实际反应动力学参数.对比图 4可知,对流出液中As(III)的拟合,初始阶段水羟锰矿体系较酸性水钠锰矿体系拟合度更高,原因是初始流入 As(III)的量少,而矿物大大过量,此时酸性水钠锰矿较水羟锰矿氧化度高,氧化能力强,矿物吸附氧化As(III)量更多.而对流出液中 As(V)的拟合,酸性水钠锰矿体系较水羟锰矿体系拟合度更高,这是因为反应生成的 As(V)和 Mn(Ⅱ)迅速吸附在矿物表面,水羟锰矿的比表面大,活性位点多,吸附量远远大于酸性水钠锰矿,因而酸性水钠锰矿拟合度较好.由此可见,通过搅拌流动法的氧化动力学模型拟合较差,是因为模型中忽略了反应过程中As(V)和Mn2+吸附的影响.

图6 合成氧化锰矿物氧化As(III)反应搅拌流动法校正动力学模型拟合Fig.6 Fitting of kinetic curves of As(III) oxidation by synthesized Mn oxide minerals with the stirred correction flow oxidation kinetic model
反应生成 As(V)一部分释放到溶液中,一部分吸附在矿物表面.研究发现,锰氧化物氧化As(III)最终矿物表面吸附的 As主要是As(V)[16-18].因此假定吸附量全部为 As(V),通过吸附量方程得到校正后的 As(V)的浓度如方程式(8)和式(9)所示,用校正后的 As(V)浓度作为新的氧化动力学曲线,并用构建的Stirred-flow的氧化动力学模型进行拟合,结果如图6所示.酸性水钠锰矿和水羟锰矿拟合度均有大幅度提高,R2分别为0.966和0.939,拟合得到的动力学速率常数分别为0.190,0.231min-1.这与Ginder-Vogel[10]采用快速X射线吸收光谱(Q-XAFS)得到的反应初始1~60s的速率常数0.282min-1基本一致,表明通过吸附量校正,用流动法氧化动力学模型拟合可得到接近于真实的化学动力学速率常数.
流动法中经过校正吸附量后实际反应生成As(V)曲线中,在初始6min阶段,加入酸性水钠锰矿溶液中 As(V)氧化率高于水羟锰矿,随着反应趋于稳定时,水羟锰矿氧化率高于酸性水钠锰矿.将化学动力学参数归一到单位比表面上,酸性水钠锰矿和水羟锰矿的速率常数分别 0.131,0.014min-1·m-2,可见,单位面积酸性水钠锰矿氧化As(III)速率远高于水羟锰矿.
2.2.3 批量实验体系 批量实验体系中,合成氧化锰氧化As(III)的动力学曲线如图7所示.反应起初 As(V)生成速率快,反应到一定时间后曲线趋于平坦,上清液中As(V)的浓度接近平衡.酸性水钠锰矿体系在60min时生成As(V)浓度趋向于稳定,氧化率为 82.2%;而水羟锰矿体系中在 180min接近稳定,其氧化率达92.2%.上清液中As(III)的浓度随着反应进行逐渐减小至平衡.酸性水钠锰矿反应体系中As(III)的去除率为85.5%,而加入水羟锰矿溶液中的去除率达97.6%.
上清液中总砷的浓度低于初始加入 500 μmol/L溶液浓度,水钠锰矿体系中总砷浓度占初始浓度97%,而水羟锰矿体系中约为80%,这说明初始阶段氧化锰矿物表面吸附有一定量的As,且水羟锰矿表面 As的吸附量高于水钠锰矿.上清液中总砷浓度在开始的20min内均有明显下降,随后慢慢上升趋于稳定,这是因为初始加入的As(III)溶液被氧化锰矿物表面所吸附,经过电子转移发生氧化还原反应生成As(V),大部分As(V)释放到溶液中,少量As(V) 吸附在了矿物表面[25].从图 7c中也可以看出,反应开始后,吸附态砷浓度一开始迅速上升,然后迅速下降逐渐趋于平衡,这是因为初始阶段表面活性位点多,而随着反应进行,反应生成的Mn2+吸附在矿物上, Mn(IV)与Mn2+反歧化作用生成活性较弱的Mn(III),造成表面活性位点钝化,此外生成的 Mn2+对 As吸附也有一定竞争影响.水羟锰矿氧化 As(III)时上清液中总砷浓度下降趋势明显高于酸性水钠锰矿,水羟锰矿吸附量远高于酸性水钠锰矿,这是因为水羟锰矿结晶弱、表面积大.

图7 合成氧化锰矿物氧化As(III)的批量法动力学曲线Fig.7 The kinetic curves of As (III) oxidation by synthesized Mn oxide minerals in batch test
层状锰氧化物有 2种反应位点,一种是矿物层间位点,一种是边面位点[27].研究表明 As(III)和As(V)主要和矿物边面位点反应而不是层间位点,Mn2+优先占据矿物层间位点,趋向形成一种三齿共角的复合物,当层间位点占据完后,Mn2+与As(III)和 As(V)竞争边面位点[28-32].锰氧化物氧化As(III)生成 Mn2+,然而上清液测定 Mn2+含量很低(图7),这说明在反应中生成的Mn2+基本被矿物完全吸附.因此,随着时间进行 As(III)反应速率降低,矿物反应性下降,除了反应产物As(V)浓度增加使逆反应速率升高之外,另一重要原因是生成的Mn2+和As(V)对矿物活性位点的钝化和屏蔽[18].
2.2.4 批量法动力学方程拟合 δ-MnO2氧化As(III) 由图8可知,采用准一级动力学方程对锰氧化物氧化As(III)的氧化反应得到的曲线进行拟合,氧化锰矿物的前5min ln(C/C0)与t呈现很好的线性关系.酸性水钠锰矿和水羟锰矿体系中 R2分别为0.996和0.972,且从回归直线的斜率可知,表观速率常数kobs分别为0.0536,0.027min-1. Ginder-Vogel等运用批量法对锰氧化物氧化As(III)的反应速率在初始 135~300s进行拟合符合准一级动力学方程,表观反应速率 0.0366min-1[10],与本实验结果基本一致.将表观速率常数归一到单位比表面上,则酸性水钠锰矿和水羟锰矿的动力学参数分别为0.021, 0.001min-1m-2,单位比表面酸性水钠锰矿氧化 As(III)速率远高于水羟锰矿.这与搅拌流动法得到趋势一致.
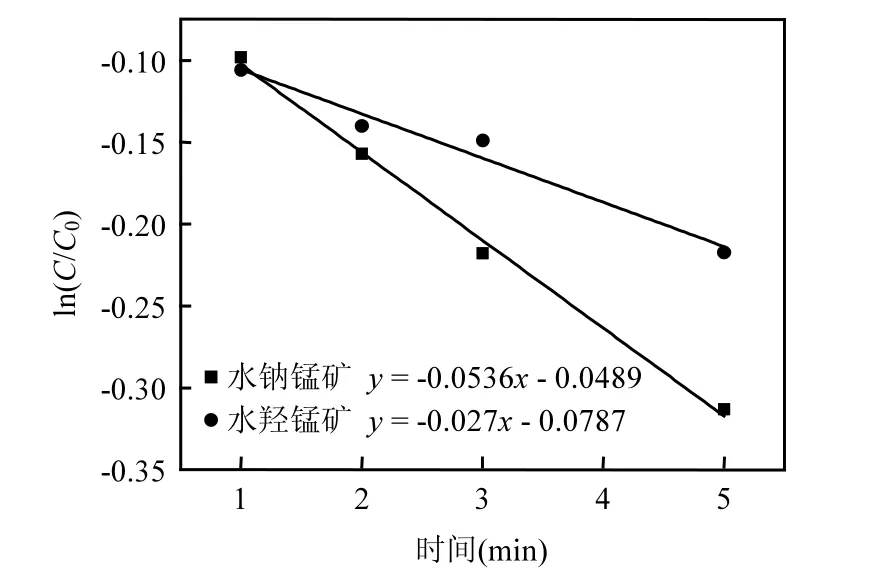
图8 合成氧化锰矿物氧化As(III)的初始5min反应的一级动力学线性拟合Fig.8 Fitting of kinetic curves of As(III) oxidation by synthesized Mn oxide minerals with the first order model
2.3 讨论
比较批量实验和流动体系拟合数据可知,批量法得到单位比表面上表观速率常数(0.021,0.001min-1·m-2)明显小于搅拌流动法得到(0.131,0.014min-1·m-2).这是因为在批量实验中,产物Mn2+和As(V)吸附在了矿物表面,屏蔽了表面活性位点,阻止了As(III)的吸附和氧化;而流动法中生成的大部分产物随流出液带出体系,大大减小了表面沉淀和逆反应的影响,得到了更接近于真实氧化反应的化学动力学参数.
在初始反应条件一致情况下,批量实验中初始阶段酸性水钠锰矿体系中氧化速率高于水羟锰矿,但反应到 120min以后,水羟锰矿对 As(III)的氧化率和去除率高于酸性水钠锰矿.相对Mn(III)位点,Mn(IV)的位点对于 As(III)和 As(V)显示了更强烈的吸附氧化能力[11,33].初始阶段虽然水钠锰矿活性位点少,但相对于矿物过量情况下,矿物表面活性位点都很充足,而水钠锰矿结晶度高、氧化度高,其结构中 Mn4+多,Mn3+和 Mn2+少[24],吸附 As(III)和释放 As(V)速率更快,因此在初始阶段其氧化速率高于水羟锰矿,归一到单位比表面上,酸性水钠锰矿和水羟锰矿表观氧化速率分别为 0.021,0.001min-1·m-2.随着反应进行,产物As(V)和 Mn2+的吸附屏蔽了矿物的反应位点,而水羟锰矿比表面积大,反应活性位点多,因此反应后期其氧化率高于酸性水钠锰矿.流动实验中校正吸附量后,初始阶段酸性水钠锰矿体系中氧化生成 As(V)高于水羟锰矿,随着反应进行(6min后),水羟锰矿体系中氧化率逐渐超过水钠锰矿,这与批量法过程所反映的动力学规律一致.Chernyshova[34]研究发现,尺寸效应对纳米氧化铁材料有较大影响,随着尺寸减小,其表面氧化催化性能减弱.水羟锰矿较酸性水钠锰矿颗粒小、易聚集,可能是其单位面积氧化速率低于酸性水钠锰矿的原因之一.总之,在反应初始阶段,相对于过量矿物来说,As(III)浓度较低,As(III)的吸附为主要反应限速步骤,而随着反应进行,矿物表面反应位点逐渐钝化或因反应而减少,反应位点数量则成为主要限速步骤.
As(V)和 Mn2+的吸附是锰氧化物钝化的主要原因.搅拌流动实验的酸性水钠锰矿体系中,随着反应进行,溶液中 As(III)浓度的逐渐增加达到稳定,此时 As(III)的去除(包括吸附氧化)速率保持稳定,同时 As(V)的浓度也基本恒定,即 As(III)氧化速率和 As(V)释放速率趋于平衡.此外,总砷浓度保持稳定,与稀释曲线接近,表明吸附态砷也处于动态平衡过程,反应过程吸附 As的量不再变化,视此时是一个稳态过程.然而,此时反应不断继续生成 As(V)和 Mn2+,其中生成的 As(V)不再吸附,而 Mn2+仍完全积累在表面,且矿物氧化速率却能保持平衡,也就是说,反应过程中的Mn2+在矿物表面上的积累在一定时间内对氧化反应速率没有影响.几种可能的解释是:(1)Mn2+转移至层间,即As(III)与表面边面位点发生反应,释放As(V)的同时,生成的 Mn2+快速释放转移至层间位点,空出下新的表面位点使位点的数量保持基本不变,维持反应速率恒定;(2)Mn2+吸附不影响As(III)的反应,即表面位点反应生成的Mn2+与表面形成一种桥键的复合物促进 As(III)的吸附[14],并与其相邻 Mn(IV)位点发生电子转移;(3)As(III)与矿物通过电化学途径进行电子转移,即As(III)首先吸附在边缘位点,失去电子发生氧化反应,电子受体不是As(III)吸附的Mn(IV, III)位点,而是把氧化锰矿物作为一个半导体传递电子,由其结构中另外活性或氧化还原电位更高的Mn(IV, III)位点提供电子,而该位点可能位于体相或层内,但因位阻效应不能吸附As(III),因此表面吸附 As (III)位点并没有价态变化,随着生成As(V)的释放可再吸附 As (III)发生反应,从而使反应速率保持不变.随着反应长时间进行,表面Mn(IV, III)位点因钝化或发生反应而不断减少,反应速率随之下降.
上述吸附量的校正方法为搅拌流动动力学模型的修正提供了思路,由于 As在氧化锰表面的吸附相对较弱,故通过吸附量校正后的模型拟合度较好,应用该法研究其它多相体系氧化还原过程,尤其是吸附强,影响大的体系还需要进一步构建反应中的吸附动力学模型或即时平衡热力学模型.土壤环境极其复杂,不仅仅是单一的静止状态或流动环境,如何结合批量法和搅拌流动法模拟土壤开放流动环境中的氧化还原过程和机制有待进一步的研究.
3 结论
3.1 建立了搅拌-流动法中氧化还原动力学模型,经过吸附量的校正后,酸性水钠锰矿、水羟锰矿在pH7下氧化As(III)动力学模型对实验数据拟合度分别为0.966和0.939,通过模型获取的反应速率常数分别为 0.190,0.231min-1,单位比表面下速率常数为 0.131,0.014min-1·m-2.单位面积酸性水钠锰矿氧化As(III)速率高于水羟锰矿.
3.2 批量实验体系中,在pH 7下得到的酸性水钠锰矿、水羟锰矿氧化As(III)的表观速率常数分别为 0.0536,0.027min-1,单位比表面上,酸性水钠锰矿和水羟锰矿表观氧化速率常数为 0.021,0.001min-1·m-2,远小于流动法得到的近似于化学动力学参数.批量法中由于产物在反应中的积累所带来的表面沉淀和逆反应而得到的表观速率常数,难以揭示化学反应动力学历程.而流动法中生成的大部分产物随流出液带出体系,大大减小了表面沉淀和逆反应的影响,能够得到更接近于真实氧化反应的化学动力学参数.
3.3 反应初始阶段,流速和搅拌速度一定条件下,As(III)的吸附为主要反应限速步骤,而随着反应的进行,由于矿物表面的反应位点因吸附As(V)、Mn2+及不断反应而减少,反应位点数量成为主要限速步骤.
[1]Smith A H, Lingas E O, Rahman M. Contamination of rinking-water by arsenic in Bangladesh: a public health emergency [J]. Bulletin of the World Health Organization,2000,78(9):1093-1103.
[2]Nordstrom D K. Worldwide occurrences of arsenic in ground water [J]. Science, 2002,296(5576):2143-2145.
[3]Duker A A, Carranza E J M, Hale M. Arsenic geochemistry and health [J]. Environmental International, 2005,31(5):631-641.
[4]Jones L C, Lafferty B J, Sparks D L. Additive and Competitive Effects of Bacteria and Mn Oxides on Arsenite Oxidation Kinetics [J]. Environmental Science and Technology, 2012,46:6548-6555.
[5]Tebo B M, Bargar J R, Clement B G, et al. Biogenic manganese oxides: Properties and mechanisms of formation [J]. Annual Review of Nuclear and Particle Science, 2004,32:287-328.
[6]Feng X H, Zhai L M, Tan W F, et al. Adsorption and redox reactions of heavy metals on synthesized Mn oxide minerals [J].Environmental Pollution, 2007,147:366-373.
[7]Feng X H, Tan W F, Liu F, et al. Oxidation of As(III) by several manganese oxide minerals in absence and presence of goethite [J].Acta Geologica Sinica-English Edition, 2006,80(2):249-256.
[8]Bar-Tal A, Sparks D L, Pesek J, et al. Kinetics of ion exchange on soil constituents using a stirred-flow chamber. I. Theoretical considerations [J]. Soil Science Society of America Journal,1990,54:1272-1277.
[9]Tournassat C, Charlet L, Bosbach D, et al. Arsenic (III) Oxidation by Birnessite and Precipitation of Manganese (II) Arsenate [J].Environmental Science and Technology, 2002,36:493-500.
[10]Ginder-Vogel M, Landrotn G, Fischel J S, et al. Quantification of rapid environmental redox processes with quick-scanning x-ray absorption spectroscopy (Q-XAS) [J]. Proceedings of the National Academy of Sciences, 2009,106(38):16124-16128.
[11]Zhu M, Paul K W, James D K, et al. Quantum chemical study of arsenic (III,V) adsorption on Mn-oxides: Implications for arsenic(III) oxidation [J]. Environmental Science and Technology, 2009,43:6655-6661.
[12]Sparks D L. Environmental Soil Chemistry [M]. 2nd Edition.Academic Press, San Diego, 2003.
[13]王代长,蒋 新,卞永荣,等.酸沉降下红壤对 K+吸附特征及反应动力学 [J]. 中国环境科学, 2003,23(1):60-64.
[14]王代长,蒋 新,卞永荣,等.模拟酸雨条件下 Cd2+在土壤及其矿物表面的解吸动力学特征 [J]. 环境科学, 2004,23(4):117-122.
[15]Lopez-Periago J E, Arias-Estevez M, Novoa-Munoz J C, et al.Copper retention kinetics in acid soils [J]. Soil Science Society of America Journal, 2006,72:63-67.
[16]Lafferty B J, Ginder-Vogel M, Sparks D L. Arsenite oxidation by a poorly crystalline manganese-oxide: 1. Stirred- flow experiments [J]. Environmental Science and Technology, 2010,44(22):8460-8466.
[17]Lafferty B J, Ginder-Vogel M, Zhu M, et al. Arsenite oxidation by a poorly crystalline manganese-oxide: 2. Results from x-ray absorption spectroscopy and x-ray diffraction [J]. Environmental Science and Technology, 2010,44(22):8467-8472.
[18]Lafferty B J, Ginder-Vogel M, Sparks D L. Arsenite oxidation by a poorly-crystalline manganese-oxide: 3. Arsenic and manganese desorption [J]. Environmental Science and Technology, 2011,45(21):9218-9223.
[19]McKenzie R M. The synthesis of birnessite, cryptomelane, and some other oxides and hydroxides of manganese [J].Mineralogical Magazine, 1971,38:493-503.
[20]Villalobos M, Toner B, Bargar J, et al. Characterization of the manganese oxide produced by pseudomonas putida strain MnBl[J]. Geochimica et Cosmochimica Acta, 2003,67(14):2649-2662.
[21]Kijima N, Yasuda H, Sato T, et al. Preparation and characterization of Open Tunnel Oxide α-MnO2Precipitated by Ozone Oxidation [J]. Journal of Solid State Chemistry, 2001,159(1):94-102.
[22]Hui Yin, Wenfeng Tan, Xionghan F, et al. Characterization of Ni-rich hexagonal birnessite and its geochemical effects on aqueous Pb2+/Zn2+and As (III) [J]. Geochimica et Cosmochimica Acta, 2012,93:47-62.
[23]Oscarson D W, Huang P M, Liaw W, The oxidation of arsenite by aquatic sediments [J]. Environmental Quality, 1980,9:700-703.
[24]Zhao Wei, Cui Haojie, Liu Fan, et al. Relationship between Pb2+Adsorption and average Mn oxidation state in synthetic birnessites [J]. Clays and Clay Minerals, 2009,57(5):513-520.
[25]Manning B A, Fendorf S E, Bostick B, et al. Arsenic(III)oxidation and arsenic(V) adsorption reactions on synthetic birnessite [J]. Environmental Science and Technology, 2002,36:976-981.
[26]Yin Hui, Feng Xionghan, Qiu Guohong, et al. Characterization of Co-doped birnessites and application for removal of lead [J].Journal of Hazardous Materials, 2011,188:341-349.
[27]Silvester E, Manceau M, Drits V A. Structure of synthetic monoclinic Na-rich birnessite and hexagonal birnessite: II.Results from chemical studies and EXAFS spectroscopy [J].American Mineralogist, 1997,82:962-978.
[28]Manceau A, Lanson M, Geoffroy N. Natural speciation of Ni, Zn,Ba, and As in ferromanganese coatings on quartz using X-ray fluorescence absorption and diffraction [J]. Geochim Cosmochim Acta, 2007,71:95-128.
[29]Feng X H, Zu Y Q, Tan W F, et al. Arsenite oxidation by several manganese oxides as affected by pH, ion strength and tartaric acid[J]. Journal of Environmental Sciences, 2006,18(2):292-298.
[30]Kwon P J, Refson K D, Bargar K, et al. Mechanisms of nickel sorption by a bacteriogenic birnessite [J]. Geochim Cosmochim Acta, 2010,74:3076-3089.
[31]Moore J N, Walker J R, Hayes T H. Reaction scheme for the oxidation of As (III) to As (V) by birnessite [J]. Clays Clay Miner,1990,38:549-555.
[32]Foster A L, Brown G E, Parks G A. X-ray absorption fine structure study of As (V) and Se (IV) sorption complexes on hydrous Mn oxides [J]. Geochim Cosmochim Acta, 2003,67(11):1937-1953.
[33]谭军凤,邱国红,刘 凡,等.Mn(III)在水钠锰矿氧化 Cr(III)反应中的作用 [J]. 环境科学, 2009,30(9):2779-2785.
[34]Chernyshova I V, Ponnurangam S, Somasundaran P. Effect of nanosize on catalytic properties of ferric (hydr)oxides in water.Mechanistic insights [J]. Journal of Catalysis, 2011,282:25-34.
Kinetic characteristics of As (III) oxidation by δ-MnO2in the simulated environment: A stirred flow study.
WANG Kun, XIA Ping-ping, LIU Fan, TAN Wen-feng, QIU Guo-hong, FENG Xiong-han*(Key Laboratory of Arable Land Conservation (Middle and Lower Reaches of Yangtze River), Ministry of Agriculture, College of Resources and Environment,Huazhong Agricultural University, Wuhan 430070, China). China Environmental Science, 2014,34(4):966~975
In this study, As (III) oxidation at the surfaces of two δ-MnO2minerals, birnessite and vernadite, was investigated using stirred-flow technique, and the corresponding model was established to describe the kinetics of the redox reactions in the stirred-flow heterogeneous system. Following correction of As adsorption, the degree of fitting of the stirred-flow kinetic data of As (III) oxidation by birnessite and vernadite using the established model was 0.980and 0.951, respectively. The obtained initial rate constants (k) per unit specific area at pH 7were 0.131 and 0.014min-1m-2,respectively, which were much greater than apparent initial rate constant (kobs), 0.021min-1m-2and 0.001min-1m-2, derived from the batch experiments. This indicated that rate constant (k) is much closer to the real chemical kintics that could be obtained using the stirred-flow technique. Both stirred-flow and the batch experiments showed that birnessite exhibited the greater reaction rate on a per surface area basis in As (III) oxidation than on vernadite, although birnessite had a relatively lower suface area and As (III) oxidation capacity. Analysis of the reaction process suggested that As (III)absorption was the rate determining step in the initial stage, and then the number of suface Mn reactive sites gradually became the rate determining step with the passivation and decrease of the sites.
As (III);δ-MnO2;stirred-flow technique;kinetics;rate constant;reactive site
X131.2
A
1000-6923(2014)04-0966-10
2013-08-12
国家自然科学基金项目(41171197和 40971142),中央高校基本科研业务费专项资金资助(2013JQ004,2010PY006)
* 责任作者, 教授, fxh73@mail.hzau.edu.cn
王 鹍(1988-),男,湖北浠水人,华中农业大学硕士研究生,研究方向为环境污染修复.
猜你喜欢
建材发展导向(2021年16期)2021-10-12
矿产综合利用(2020年1期)2020-07-24
矿产综合利用(2020年1期)2020-07-24
矿产综合利用(2020年1期)2020-07-24
中国锰业(2019年3期)2019-07-11
福建基础教育研究(2019年8期)2019-05-28
消费导刊(2018年8期)2018-05-25
中成药(2018年2期)2018-05-09
中学生数理化·高二版(2016年3期)2016-12-26
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18
