
奥硝唑片人体生物等效性研究
2014-06-05 15:31路文斌
中国医药指南 2014年29期
路文斌
(中国人民解放军第86医院,安徽 马鞍山 243100)
奥硝唑片人体生物等效性研究
路文斌
(中国人民解放军第86医院,安徽 马鞍山 243100)
目的比较口服奥硝唑片试验制剂与国产上市制剂的药代动力学参数,进行生物利用度和生物等效性评价。方法开放、随机、单次给药、双周期交叉临床研究。共20例健康受试者。结果试验制剂和参比制剂的药代动力学结果:T1/2分别为(17.010±2.682)h和(17.446 ±2.519)h;Tmax分别为(1.550±0.759)h和(1.250±0.526)h;Cmax分别为(9.476±1.083)μg/μL和(9.592±1.251)μg/μL;AUC0-t分别(225.604 ±44.327)μg/(μL·h)和(213.880±43.834)μg/(μL·h);AUC0-∞分别为(231.269±48.014)μg/(μL·h)和(219.655±46.518)μg/(μL·h)。试验制剂对于参比制剂的平均相对生物利用度F值(106.9±15.4)%。两种制剂的AUC0-t、AUC0-∞及Cmax经对数转换后双单侧t检验结果P<0.05,接受两种制剂生物等效的假设。90%置信区间的计算结果:Cmax为93.7%~104.5%,AUC0-t为100.0%~112.1%,AUC0-∞为99.7%~111.9%,按照生物等效性判定标准,可判定两种制剂生物等效。结论两制剂间无显著性差异,两制剂具有生物等效性。适用与Ⅰ期临床研究。
奥硝唑;生物等效性;生物利用度;HPLC
奥硝唑是继甲硝唑、替硝唑之后第3代硝基咪唑类衍生物,20世纪70年代上市以来,由于其良好的抗厌氧菌和抗原生质(如滴虫等)感染作用,临床应用日趋广泛。奥硝唑进入易感的微生物细胞后,在无氧或少氧环境和较低的氧化还原电位下,其硝基易被电子传递蛋白还原成具细胞毒作用的氨基,抑制细胞DNA的合成,并使已合成的DNA降解,破坏DNA的双螺旋结构或阻断其转录复制,从而使病原体细胞死亡[1]。本研究目的是观察口服奥硝唑片后的血药浓度经时过程,估算相应的药代动力学参数,并以国产上市制剂为参比制剂,进行生物利用度和生物等效性评价,为临床申报提供数据[2]。
1 材料与方法
1.1 药品、试剂及仪器
①药品。试验制剂:奥硝唑片,规格:0.25克/片;批号:090417201;参比制剂:奥硝唑片,规格:0.25克/片;批号:1005416032;对照品:奥硝唑对照品(中国药品生物制品检定所,批号:100608-200301)。甲硝唑对照品(内标,中国药品生物制品检定所,批号:100191-200305)。②试剂。甲醇、乙腈(TEDIA公司),色谱纯;冰醋酸(上海化学试剂有限公司),分析纯;无水乙醚(上海中试化工总公司),分析纯;水,自制超纯水。③仪器。LC-10ATvp高效液相色谱仪,SPD-10Avp紫外检测器,LC-10ATvp输液泵,Cs-light色谱数据工作站软件,均为日本岛津公司生产;AT-130柱温箱,AUTO SCIENCE公司生产;XW-80A微型旋涡混合仪(上海沪西分析仪器厂);XS105分析天平(梅特勒-托利多仪器有限公司);16K高速离心机(珠海黑马医学仪器有限公司);Milli-Q Academic纯水机(密理博(上海)贸易有限公司);HGC-24型氮吹仪(北京瑞邦兴业科技有限公司)。
1.2 方法
1.2.1 奥硝唑测定的HPLC测定方法
色谱柱:岛津VP-ODS 150 mm×4.6 mm,流速:1.0 μL/min;流动相:乙腈∶水(含0.1%冰醋酸)=25∶75(V∶V);灵敏度:0.001AUFS;检测波长:316 nm;柱温:40 ℃,进样量:20μL。
1.2.2 奥硝唑测定的HPLC的方法学验证
1.2.2.1 奥硝唑血浆样品处理方法:精密量取10 μL甲硝唑内标溶液(10.23 μg/μL)置于空白离心管中,加入100 μL血浆样品,涡旋30 s,精密加入1 μL乙醚,涡旋3 min,14000 rpm离心10 min,取有机层,N2下吹干,200 μL流动相复溶,涡旋混匀,14000 rpm离心5 min,20 μL进样。
1.2.2.2 方法的专属性:精密取6份不同来源空白血浆各100 μL,按
1.2.2.1项下操作;同时混合该6份空白血浆,取100 μL,按1.2.2.1项下操作;取奥硝唑标准溶液(13.350 μg/μL)和内标溶液(10.23 μg/μL)各10 μL,N2下吹干,加入200 μL流动相,混匀后20 μL进样;取奥硝唑标准溶液(13.350 μg/μL)和甲硝唑内标溶液(10.23 μg/μL)各10 μL,N2下吹干,加入100 μL空白血浆,按1.2.2.1项下操作;取1号受试者第一周期口服受试制剂24 h后收集的血浆样品,按1.2.2.1项下操作。
1.2.2.3 血浆中奥硝唑标准曲线:精密量取10 μL甲硝唑内标溶液(10.23 μg/μL)于空白离心管中,再加入不同浓度的奥硝唑标准溶液各10 μL,N2下吹干,加入100 μL空白血浆配制成药物浓度分别为0.083、0.334、0.668、2.670、5.340、10.680和21.360 μg/μL的样品,按“血浆样品处理方法”项下操作,进样分析,建立奥硝唑的标准曲线。以待测物浓度(x)为横坐标,待测物(As)与内标物(Ai)的峰面积比值(y,As/Ai)为纵坐标,用加权法(W=1/χ2)进行回归运算,求得的直线方程即为标准曲线。
标准曲线各浓度点的实测值与标示值之间的偏差在可接受的范围之内时,可判定标准曲线合格。可接受范围一般规定为最低浓度点的偏差在±20%以内,其余浓度点的偏差在±15%以内。
1.2.2.4 血浆中奥硝唑定量下限:取空白离心管,加入甲硝唑内标溶液(10.23 μg/μL)和0.834 μg/μL奥硝唑标准溶液各10 μL,N2下吹干,再加入100 μL空白血浆配制成血浆中药物浓度为0.083 μg/μL的样品,共5份,按1.2.2.1项下操作。每个浓度取5个样本进行分析,根据伴随的标准曲线,计算定量下限(LLOQ)样品的浓度。其准确度应在真实浓度的80%~120%范围内,相对标准差(RSD)应<20%。
1.2.2.5 血浆中奥硝唑精密度与准确度:取空白离心管加入甲硝唑内标溶液(10.23 μg/μL)10 μL,再精密加入不同浓度的奥硝唑QC溶液各10 μL,N2下吹干,加入100 μL空白血浆,配制成奥硝唑低、中、高三个浓度(分别为0.167、1.335和17.088 μg/μL)的样品,按1.2.2.1项下操作。在同一分析批内和连续3个分析批间对奥硝唑的质控样本进行处理分析,测定质控样品的批内精密度与准确度。低、中、高浓度各6个样本进行分析,根据伴随的标准曲线,计算质控样品的浓度。准确度一般应在85%~115%范围内(一般偏差应<15%),在LLOQ附近应在80%~120%范围内。

图1A 不同来源混合空白血浆色谱图

图1B 奥硝唑(13.350 μg/μL)和甲硝唑(10.23 μg/μL)标准溶液加入流动相后色谱图

图1C 奥硝唑(1.335 μg/μL)和甲硝唑(1.023 μg/μL)血浆样品色谱图

图1D 1号受试者第一周期口服受试制剂24 h后收集的血浆中奥硝唑样品色谱图

图2 定量下限图谱(LLOQ-1)
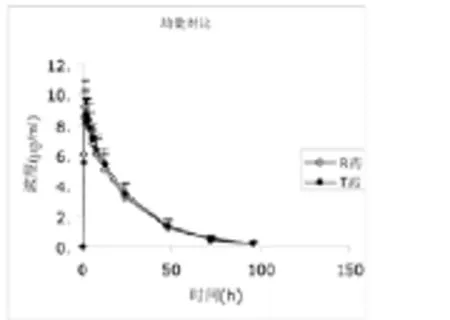
图3 20名受试者口服奥硝唑的均数对比图
1.2.2.6 血浆中奥硝唑提取回收率:取空白离心管,分别加入甲硝唑内标溶液(10.23 μg/μL)和不同浓度奥硝唑QC溶液各10 μL,N2下吹干,加入100 μL空白血浆配制成低、中、高三个浓度(分别为0.167、1.335和17.088 μg/μL)的样品各3份,按1.2.2.1项下操作,每个浓度各3个样本进行分析,得到目标分析物的峰面积为A;取空白大鼠、Beagle犬和人肝微粒体各100 μL,将空白样品进行预处理后获得提取液,以此提取液制备低、中、高浓度的QC样品,每个浓度各3个样本进行分析,得到目标分析物峰面积为B;取奥硝唑和内标各10 µL,混匀后加入流动相至100 µL,每个浓度各3个样本进行分析,得到目标分析物平均峰面积为C;依“A/C×100%”计算提取回收率,依“B/C×100%”计算介质效应。
1.2.2.7 血浆中奥硝唑稳定性:分别考察血浆样品处理后室温放置稳定性,血浆样品室温放置再处理稳定性,血浆样品反复冻融3次稳定性,血浆样品长期冷冻保存条件下的稳定性。如下:取空白离心管精密加入甲硝唑内标溶液(10.23 μg/μL)和不同浓度的奥硝唑的QC溶液各10 μL,N2下吹干,加入100 μL空白血浆,配制成奥硝唑低、高两个浓度(0.167和17.088 μg/μL)的样品,各九份,每份三样本分析。三份按1.2.2.1项下操作。在室温下放置0、4 、23 h后进样分析,考察已处理样品在室温条件下的稳定性;一份在室温下放置6 h后,按1.2.2.1项下操作。进样分析,考察药物在血浆中的室温放置再处理稳定性;三份在-20 ℃冰箱,反复冻融3次,每解冻一次后,按1.2.2.1项下操作。进样分析,考察血浆样本在反复冻融条件下的稳定性;两份在-20 ℃冰箱分别冷冻7 、69 d后,按1.2.2.1项下操作,进样分析,考察血浆样本在长期冷冻条件下的稳定性。和0 h相比,不同时间段样品RSD<15%,可认为稳定性良好。
1.2.3 奥硝唑样本测定过程中的随行标准曲线及质量控制
为减少系统误差,在进行样品分析时,每批建立一条随行标准曲线计算血药浓度,并测定质控样品(奥硝唑低、中、高浓度分别为:0.167、1.335和17.088 μg/μL),确保计算得到的药物浓度准确可靠,操作步骤同1.2.2.3和1.2.2.5,标准曲线各浓度点的实测值与标示值之间的偏差在可接受的范围之内时,可判定标准曲线合格。可接受范围一般规定为最低浓度点的偏差在±20%以内,其余浓度点的偏差在±15%以内。只有合格的标准曲线才能对临床待测样品进行定量计算。
每个分析批生物样品测定时应建立新的标准曲线,并随行测定高、中、低三个浓度的质控样品。每个浓度至少双样本,并应均匀分布在未知样品测试顺序中。当一个分析批中未知样品数目较多时,应增加各浓度质控样品数,使质控样品数大于未知样品总数的5%。质控样品测定结果的偏差一般应<15%,低浓度点偏差一般应<20%。最多允许1/3的质控样品结果超限,但不能出现在同一浓度质控样品中。如质控样品测定结果不符合上述要求,则该分析批样品测试结果作废。
1.2.4 奥硝唑样本采集和测定
采用双周期自身交叉对照设计,将入选的20名健康受试者,随机分为A组和B组,分别口服受试制剂(奥硝唑片,0.25克/片×2片)和参比制剂(奥硝唑片,0.25克/片×2片),试验间的清洗期为2周。
受试者于试验当日早晨单次空腹口服相应药物0.5 g,用200 μL温开水送服,分别于服药前(0 h)和服药后0.5、1.0、1.5、2.0、2.5、3.0、4.0、6.0、8.0、12.0、24.0、48.0、72.0、96.0 h从受试者静脉前臂静脉采血4 μL,置试管中,离心,取血浆于-20 ℃冰箱储存,用于血药浓度检测。试验间的清洗期为2周,方法同前。服药后2 h可饮水,4 h统一进清淡饮食,该研究在符合条件的GCP试验室完成[3]。
精密量取10 μL甲硝唑内标溶液(10.23 μg/μL)置于空白离心管中,分别加入1~20号受试者两周期0、0.5、1.0、1.5、2.0、2.5、3.0、4.0、6.0、8.0、12.0、24.0、48.0、72.0、96.0 h时间点血浆样品100 μL,按1.2.2.1项下操作。
1.2.5 统计学处理
2 结 果
2.1 奥硝唑测定的方法学结果
2.1.1 专属性
结果见图1(A~D),显示奥硝唑保留时间约为5.7 min;内标甲硝唑的保留时间约为2.9 min。结果表明空白血浆中内源性物质不干扰奥硝唑和内标的测定。
2.1.2 标准曲线
从表1可以看出,所考察的三条标准曲线相关系数r均>0.990,标准曲线范围内线性关系良好。

表1 血浆中奥硝唑三天标准曲线峰面积比、标准曲线方程及相关系数
2.1.3 定量下限
以标准曲线的最低点作为定量下限,五样本的分析结果可见奥硝唑定量下限偏差在15%范围内,RSD为8.23%,符合相关技术指导原则要求。奥硝唑血样定量下限为0.083 μg/μL。结果见表2和图2。
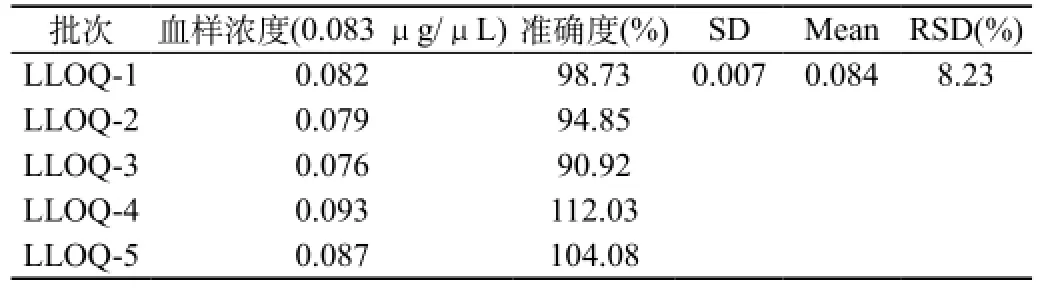
表2 奥硝唑定量下限结果
2.1.4 准确度与精密度
由表3可见,与配制浓度相比,各样品的批内的相对标准差(RSD)均<15%。结果表明精密度与准确度均符合生物样本检测方法的要求。低、中、高三个浓度(0.167、1.335和17.088 μg/μL)批内和批间的相对标准差(RSD)均<10%。
2.1.5 回收率
回收率结果见表4。奥硝唑提取回收率为67.51%~84.00%,内标甲硝唑提取回收率为58.27%~74.18%,药物及内标回收率稳定,说明该分析方法满足检测要求。
2.1.6 稳定性考察
由表5(A、B)可见,和0 h相比,各个时间段RSD均<15%。结果表明奥硝唑血浆样本室温下6 h内稳定性较好,血浆样本处理后溶液23 h稳定性较好;反复冻融3次以及长期冷冻69 d稳定性均较好。
2.1.7 奥硝唑样本测定过程中的随行标准曲线及质量控制
伴随标准曲线及质控测定结果显示:伴随标准曲线线性关系良好,质控样品偏差在15%范围内,RSD<10%,血药浓度数据测定准确,结果可靠。结果见表6、7。
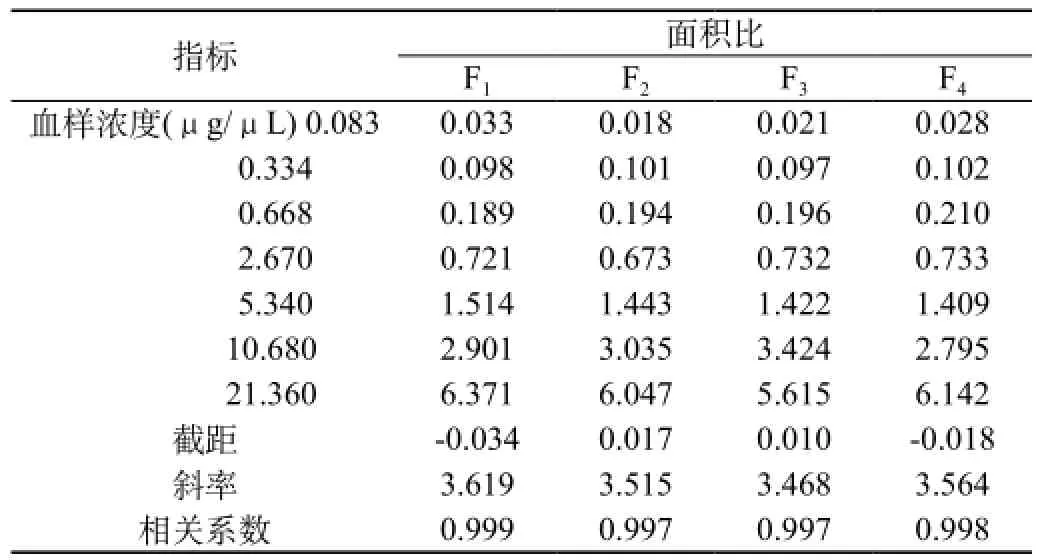
表6 血浆中奥硝唑随行标准曲线峰面积比、曲线方程及相关系数

表7 奥硝唑质控样品测定浓度及其均数、标准差
2.1.8 奥硝唑血浆样本测定结果
20名受试者口服受试制剂和参比制剂,血浆中奥硝唑血药浓度-时间数据见表8、9。
2.1.9 血药浓度-时间曲线
20名健康受试者口服受试制剂和参比制剂后奥硝唑的均数对比图,见图3。其中T表示受试制剂,R表示参比制剂。
2.2 统计学分析(生物等效性评价)
2.2.1 药代动力学参数
Cmax和Tmax由实测数据直接表示。结果见表10、11。
2.2.2 方差分析
受试制剂和参比制剂两制剂的ln(AUC0-96)、ln(AUC0-∞)、ln(Cmax)方差分析结果见表12A、表12B、表12C、表12D、表12E、表12F。结果表明ln(AUC0-96)、ln(AUC0-∞)、ln(Cmax)在药剂间、周期间差异均无统计意义,在个体间差异有统计意义(P<0.05)。
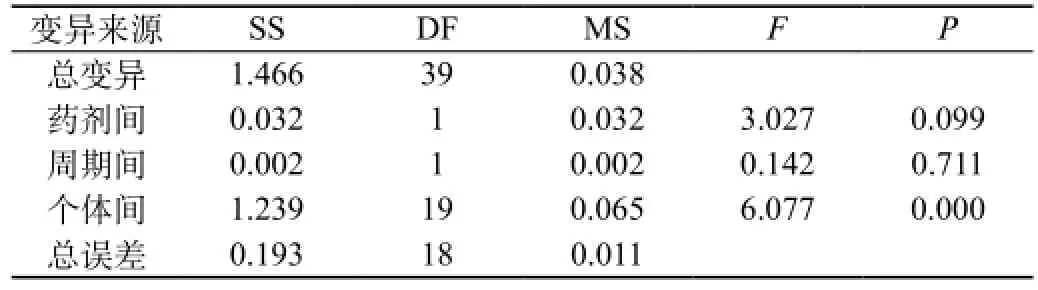
表12A 两制剂ln(AUC0-96)方差分析结果
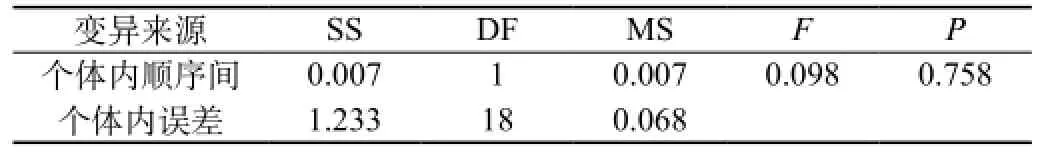
表12B 两制剂ln(AUC0-96)个体间变异方差分析结果
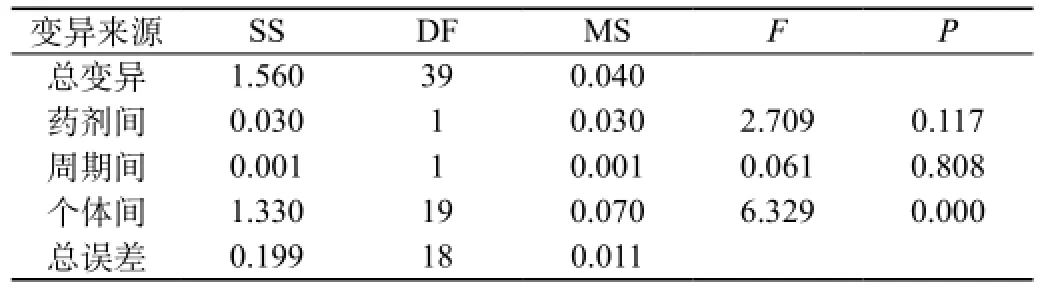
表12C 两制剂ln(AUC0-∞)方差分析结果
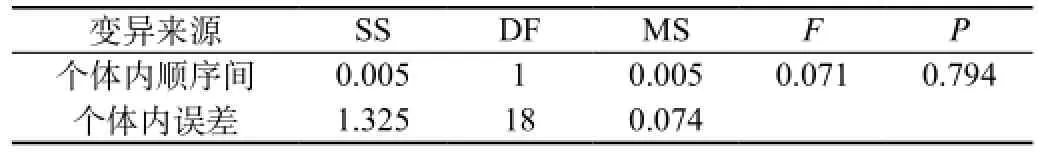
表12D 两制剂ln(AUC0-∞)个体间变异方差分析结果
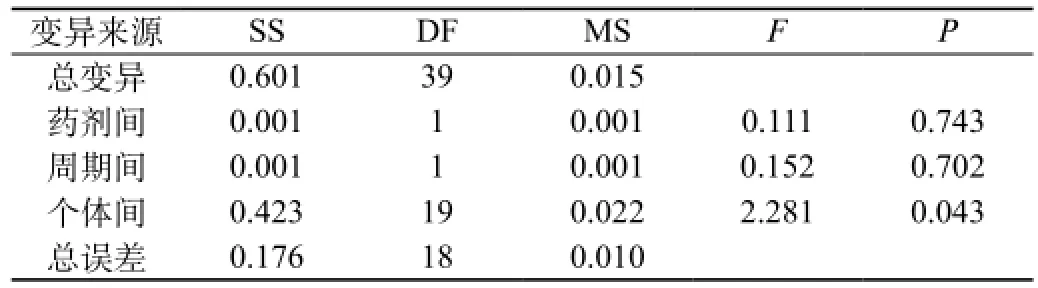
表12E 两制剂ln(Cmax)方差分析结果

表12F 两制剂ln(Cmax)个体间变异方差分析结果
2.2.3 双向单侧t检验及[1-2α]置信区间分析
ln(AUC0-96)、ln(AUC0-∞)、ln(Cmax)双向单侧t检验结果及[1-2α]置信区间分析见表13、表14、表15。表13、表14结果表明两种制剂的药代动力学参数ln(AUC0-96)、ln(AUC0-∞)的H0不成立,而接受H1,同时结果表明ln(AUC0-96)、ln(AUC0-∞)的[1-2α]置信区间在80%~125%范围内,说明两种制剂在吸收程度上具有生物等效性。表15结果表明两种制剂的药代动力学参数ln(Cmax)的H0不成立,而接受H1,同时结果表明ln(Cmax)的[1-2α]置信区间在75%~133%范围内,说明两制剂在达峰浓度上具有生物等效性。
2.2.4 Tmax的非参数检验
Tmax的非参数检验,结果表明Tmax的药剂间差异无统计意义(P>0.05)。见表16。
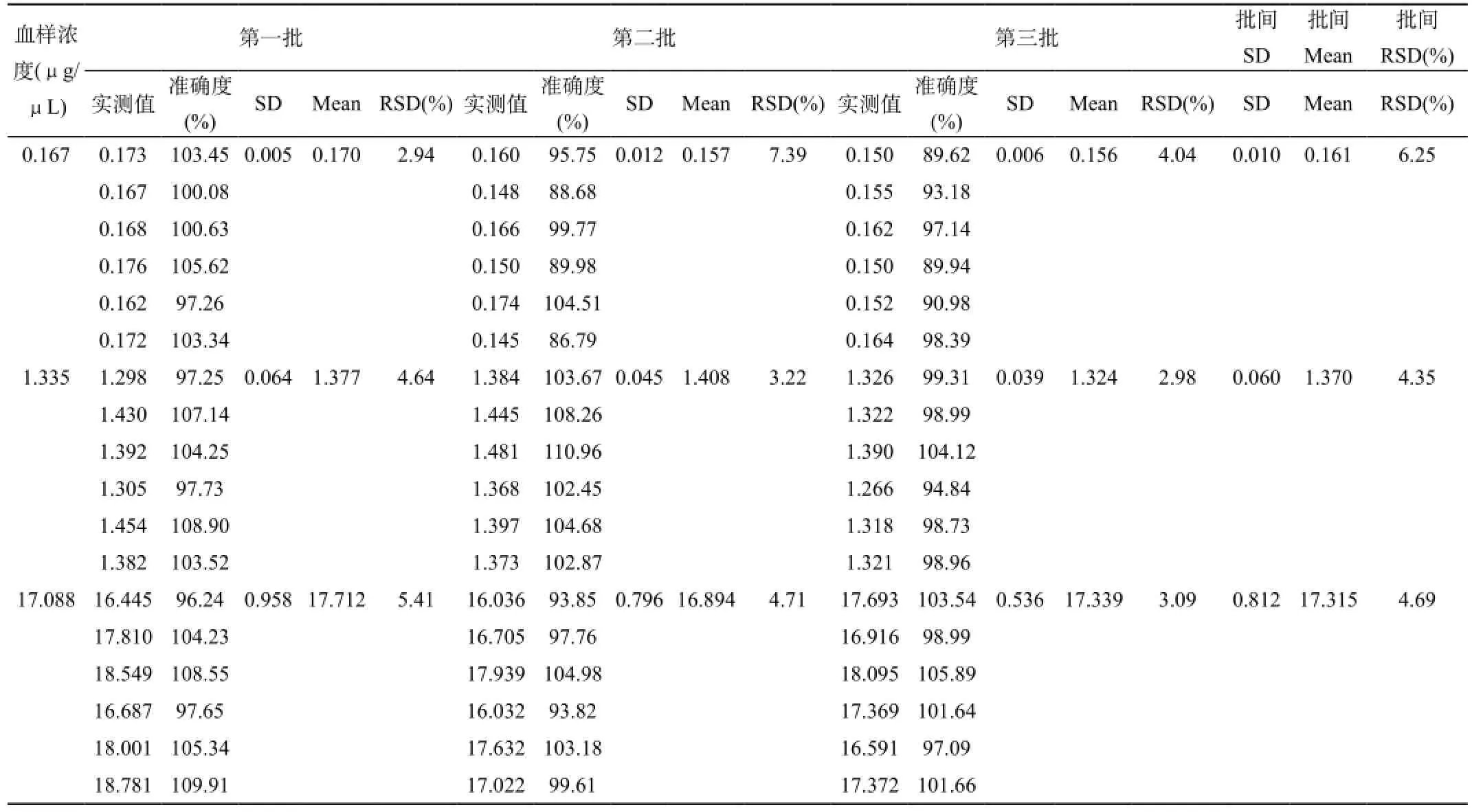
表3 奥硝唑精密度与准确度试验结果

表4 血浆中奥硝唑和内标甲硝唑提取回收率结果
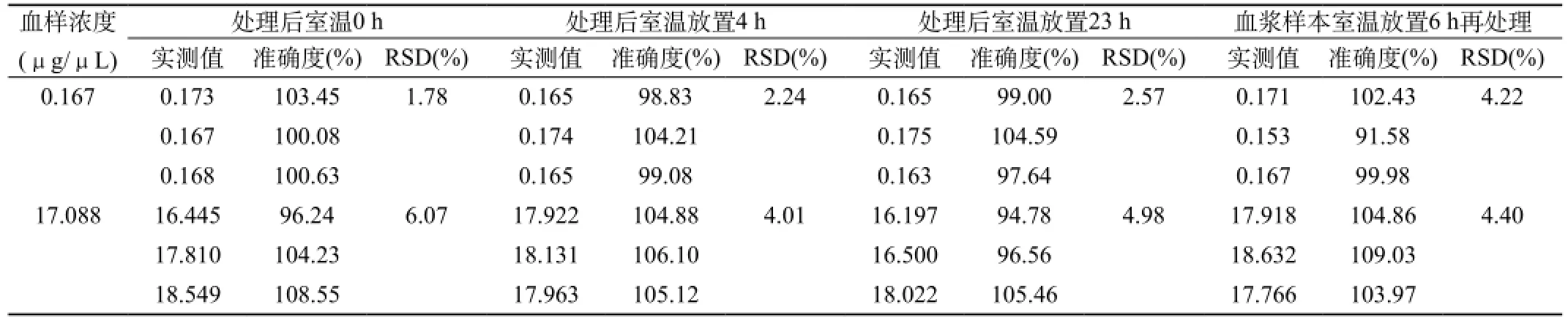
表5A 血浆中奥硝唑样品的室温稳定性及其处理液稳定性试验结果
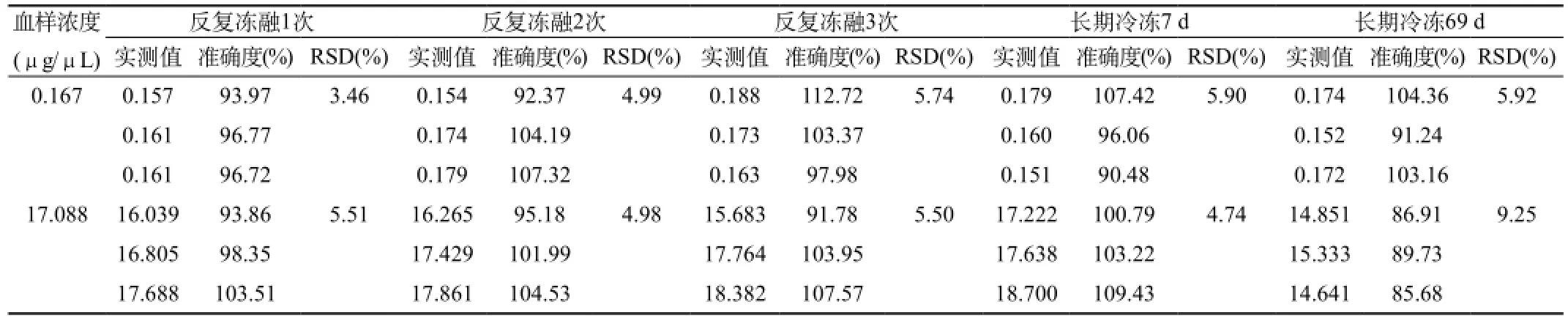
表5B 血浆中奥硝唑样品的冻融稳定性及长期冷冻稳定性试验结果

表8 20名受试者口服受试制剂后血浆中奥硝唑的浓度-时间数据(μg/μL)
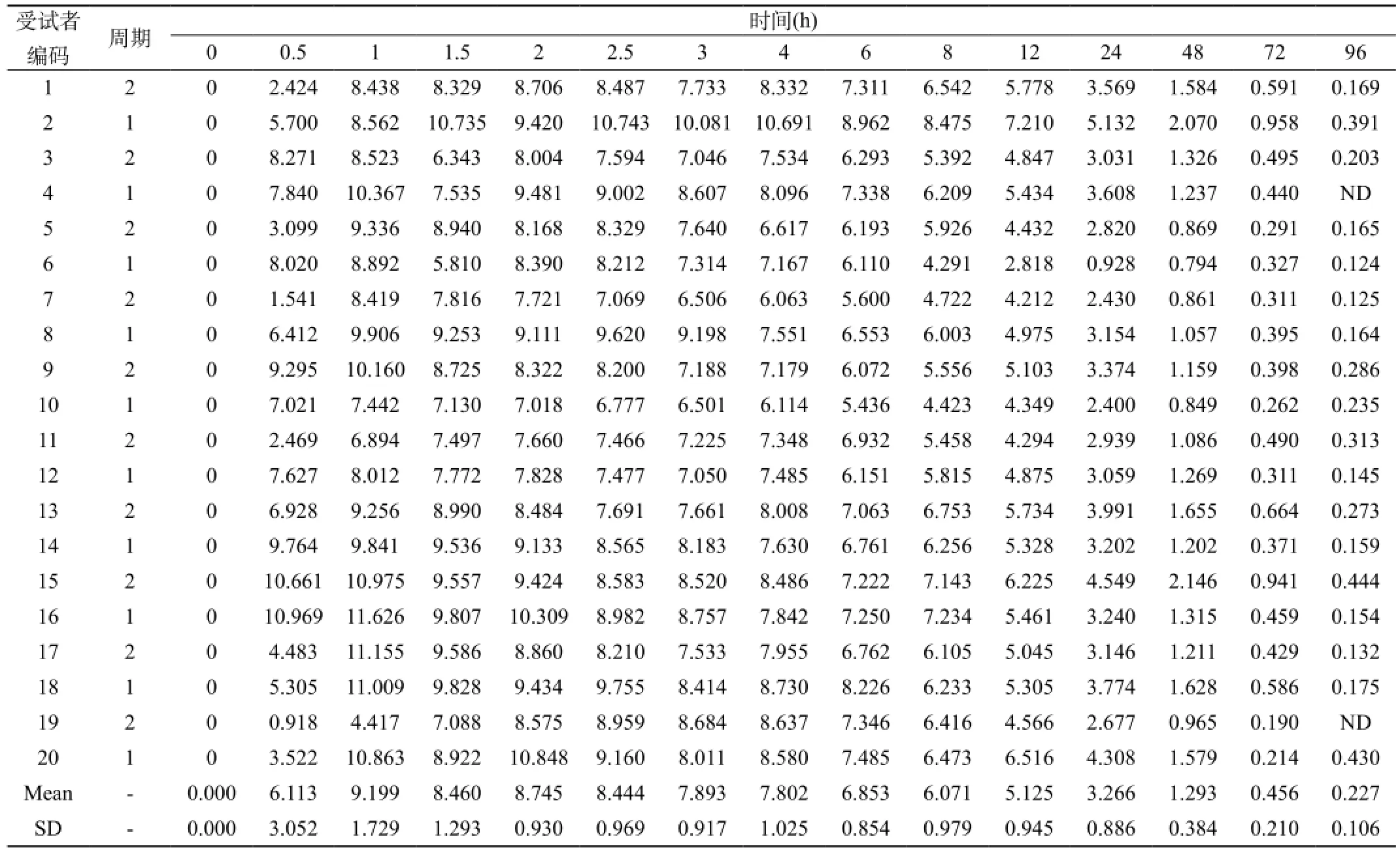
表9 20名受试者口服参比制剂后血浆中奥硝唑的浓度-时间数据(μg/μL)
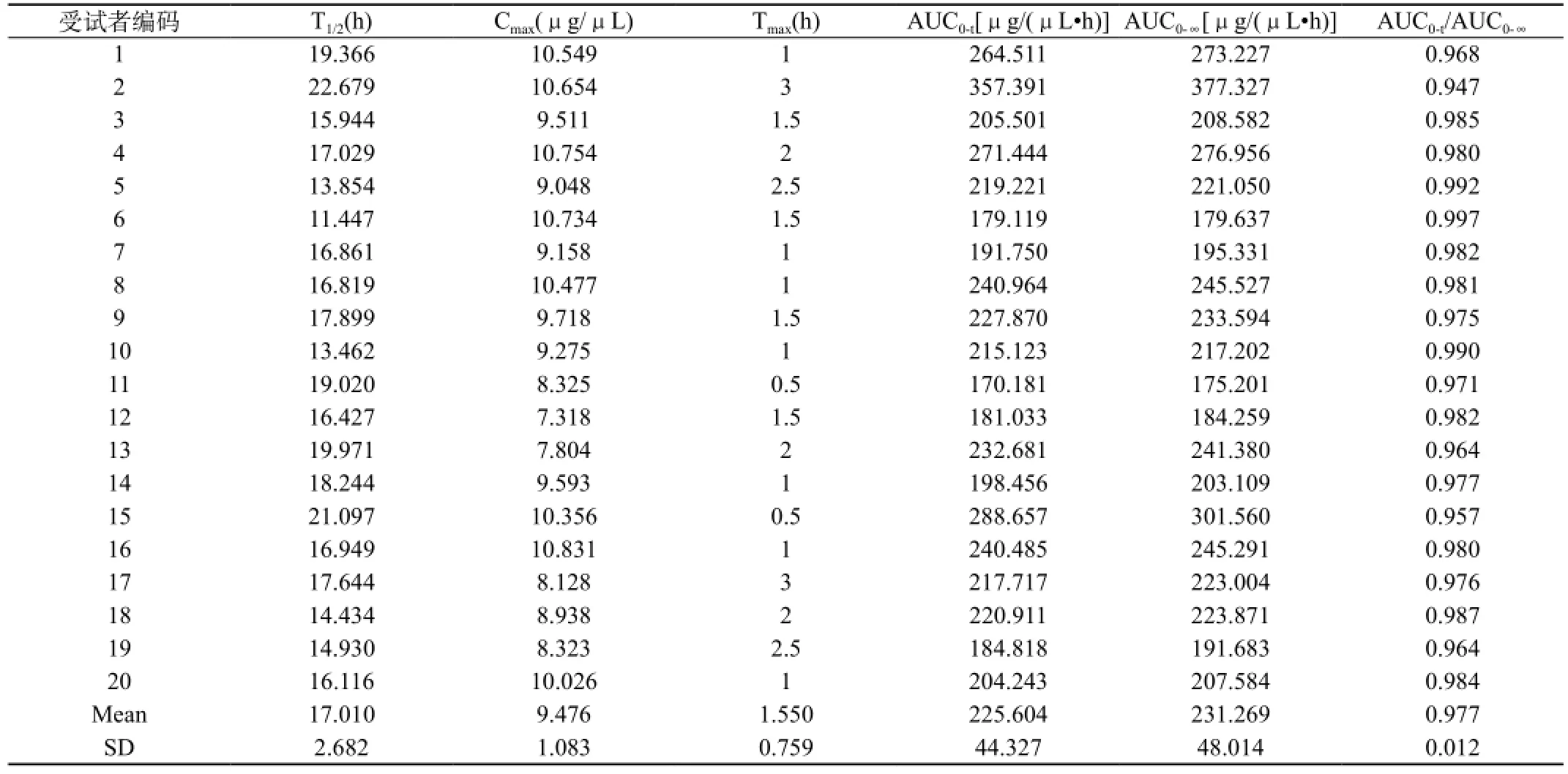
表10 20名受试者口服受试制剂后奥硝唑的药代动力学参数(T)

表11 20名受试者口服参比制剂后奥硝唑的药代动力学参数(R)
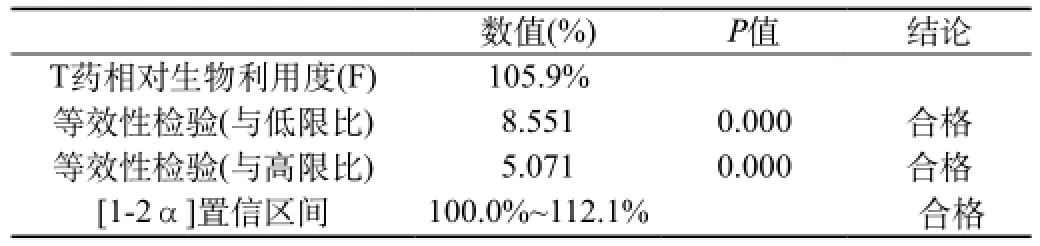
表13 ln(AUC0-96)等效性分析(ABE):双向单侧t检验(T:R)

表14 ln(AUC0-∞)等效性分析(ABE):[1-2α]置信区间法(T:R)

表15 ln(Cmax)等效性分析(ABE):双向单侧t检验(T:R)

表16 非参数检验结果
3 结论与讨论
本文采用HPLC内标法测定健康受试者血浆中奥硝唑浓度,血浆中内源性物质不干扰样品的测定,奥硝唑标准曲线线性范围为:0.083~21.360 μg/μL,定量下限为0.083 μg/μL(SN>10);血浆中奥硝唑提取回收率为67.51%~84.00%,内标甲硝唑提取回收率为58.27%~74.18%;批内和批间RSD均<10%。该方法符合生物样品分析要求。
经药代动力学参数计算,受试制剂的T1/2(17.010±2.682)h、Cmax(9.476±1.083)μg/μL、Tmax(1.550±0.759)h、AUC0-96(225.604±44.327)μg/(μL·h)、AUC0-∞(231.269±48.014)μg/ (μL·h),参比制剂的T1/2(17.446±2.519)h、Cmax(9.592±1.251)μg/μL、Tmax(1.250±0.526)h、AUC0-96(213.880±43.834)μg/ (μL·h)、AUC0-∞(219.655±46.518)μg/(μL·h)。
用面积法估算试验制剂的相对生物利用度为(106.9±15.4)%,符合生物利用度要求。
受试制剂和参比制剂中奥硝唑ln(AUC0-96)、ln(AUC0-∞)经方差分析,两制剂间无显著性差异;经双向单侧t检验,两制剂[1-2α]置信区间分别为100.0%~112.1%、99.7%~111.9%,在80%~125%范围内,表明两制剂在吸收程度上具有生物等效性。ln(Cmax)经双向单侧t检验,两制剂[1-2α]置信区间93.7%~104.5%,在75%~133%范围内,表明两制剂在达峰浓度上具有生物等效性。Tmax采用非参数检验,两制剂间无显著性差异,表明两制剂在达峰时间上具有生物等效性。
目前已发表文献与本试验均采用HPLC法测定奥硝唑浓度,文献中服药剂量为1.5 g,采用血样酸化后乙酸乙酯两次提取法测定血药浓度,而本试验服药剂量为0.5 g,采用乙醚一次提取法测定血药浓度,血样处理方法更为简单[4]。
本试验中受试制剂和参比制剂的各药代参数数据与参考文献相比并无统计学差异;数据的绝对值差异(非统计学差异)与实验室、检测系统、受试对象以及参比制剂批次不同等因素有关。因此,本研究中受试制剂和参比制剂的药代动力学参数与文献报道基本一致[5-8]。整个试验过程中,20名受试者均表现出高度的依从性,严格遵循试验方案,无漏采血样、漏服药现象出现,没有出现中途脱落者。本研究可为奥硝唑临床申报提供参考。
[1] 奥硝唑片研究者手册[S].
[2] GCL2-1.《化学药物制剂人体生物利用度和生物等效性研究技术指导原则》指导原则编号[H].
[3] 国家食品药品监督管理局.药物临床试验质量管理规范[S].2003.
[4] 李芹,王本杰,郭瑞臣.奥硝唑胶囊人体药代动力学及生物等效性研究[J].中国临床药理学杂志,2002,20(2):119-121.
[5] 温清,李俊生,董瑞谦,等.奥硝唑片、奥硝唑胶囊人体生物等效性研究[J].中国药师,2003,6(5):259-260.
[6] 魏敏吉,李天云,赵彩芸,等.奥硝唑片在健康人体的生物等效性[J].中国临床药理学杂志,2010,28(11):3327-3332.
[7] 李芹,陈建忠,郭瑞臣.奥硝唑血药浓度的高效液相色谱法测定及国产和进口片剂的药动学和生物等效性研究[J].中国现代应用药学杂志,2004,21(2):129-132.
[8] 汝玲,陈汇,庞雪冰,等.高效液相色谱法测定奥硝唑的血药浓度及其人体药动学研究[J].中国新药杂志,2004,13(11):1027-1029.
R969
B
1671-8194(2014)29-0062-07
猜你喜欢
中国典型病例大全(2022年13期)2022-05-10
锦州医科大学报(2022年2期)2022-05-07
浙江化工(2022年1期)2022-02-19
口腔护理用品工业(2021年4期)2021-11-02
口腔护理用品工业(2021年4期)2021-11-02
皮肤病与性病(2021年3期)2021-07-30
四川畜牧兽医(2020年7期)2020-07-20
饮食保健(2019年16期)2019-01-12
中成药(2018年6期)2018-07-11
中国信息化·学术版(2013年3期)2013-06-25
