
Production of Bio-gasoline by Co-cracking of Acetic Acid in Bio-oil and Ethanol*
2014-07-18 11:56WANGShurong王树荣WANGYurong王誉蓉CAIQinjie蔡勤杰andGUOZuogang郭祚刚
WANG Shurong (王树荣)**, WANG Yurong (王誉蓉), CAI Qinjie (蔡勤杰) and GUO Zuogang (郭祚刚)
State Key Laboratory of Clean Energy Utilization, Zhejiang University, Hangzhou 310027, China
Production of Bio-gasoline by Co-cracking of Acetic Acid in Bio-oil and Ethanol*
WANG Shurong (王树荣)**, WANG Yurong (王誉蓉), CAI Qinjie (蔡勤杰) and GUO Zuogang (郭祚刚)
State Key Laboratory of Clean Energy Utilization, Zhejiang University, Hangzhou 310027, China
Acetic acid was selected as the model compound representing the carboxylic acids present in bio-oil. This work focuses the co-cracking of acetic acid with ethanol for bio-gasoline production. The influences of reaction temperature and pressure on the conversion of reactants as well as the selectivity and composition of the crude gasoline phase were investigated. It was found that increasing reaction temperature benefited the conversion of reactants and pressurized cracking produced a higher crude gasoline yield. At 400 °C and 1 MPa, the conversion of the reactants reached over 99% and the selectivity of the gasoline phase reached 42.79% (by mass). The gasoline phase shows outstanding quality, with a hydrocarbon content of 100%.
bio-oil, co-cracking, acetic acid, ethanol, hydrocarbons
1 INTRODUCTION
Biomass is a clean and renewable energy. Bio-oil, obtained from fast pyrolysis of biomass, is considered to be a potential substitute for conventional transport fuels (gasoline and diesel) [1-3]. However, the application of bio-oil is limited due to the drawbacks of crude bio-oil, such as high water and oxygen content, high viscosity, corrosiveness and low heating value. In order to overcome these disadvantages, researchers have tried to use catalytic cracking to upgrade crude bio-oil and convert it to high-grade hydrocarbon fuels [4-6].
Bio-oil has a complicated composition and is rich in oxygenated components, such as carboxylic acids, aldehydes, ketones, alcohols, esters, ethers and sugars [7, 8]. The cracking behavior observed for different model compounds of bio-oil has shown that, although most components of bio-oil have the potential to be converted into liquid hydrocarbon fuels, the unfortunate catalyst deactivation problems must be solved [9-11]. Due to the influence of oxygen content and unsaturation degree on bio-oil cracking, researchers have introduced the effective hydrogen-carbon ratio (H/C)effto evaluate the cracking ability of compounds in bio-oil, given as [4, 12].

Mentzel and Holm tested the lifetime of catalysts in the cracking of model bio-oil compounds with different effective hydrogen-carbon ratios and demonstrated that the catalyst deactivation occurred more easily for chemicals with low value of (H/C)eff[12].
Crude bio-oil has a low value of (H/C)eff, and researchers have tried to promote it by using chemicals with relatively high (H/C)effas diluents. Ethanol is an ideal solvent with (H/C)effof 2, which makes it more stable in the conversion process under similar operating conditions. Meanwhile, alcohols have good solubility and can serve as a solvent for bio-oil. At present, ethanol can be produced on a large scale from biomass fermentation [13]. The upgraded products resulted from the co-cracking of ethanol and bio-oil have similar physicochemical properties to gasoline, which can be used as commercial gasoline additives.
Carboxylic acid is one of the main chemical families present in bio-oil, and represents the main reason for bio-oil corrosiveness. Acetic acid is the predominant carboxylic acid in bio-oil and results from the decomposition of cellulose and hemi-cellulose [14, 15]. Demirbas [16] quantified the amount of acetic acid in several bio-oils, and found that it could reach 16.8% of biomass dry mass. Moreover, molecular distillation of bio-oil can produce bio-oil fraction with high acetic acid content [17-19]. Therefore, the effective conversion of acetic acid to hydrocarbons has considerable significance. However, the effective H/C of acetic acid is 0 and carbon deposits form very easily in the cracking of acetic acid, deactivating the catalyst. One effective way to co-crack acetic acid and ethanol may be to improve the stability of the catalyst. In our early research, we studied the co-cracking of cyclopentanone and methanol, and obtained high selectivity and a high quality gasoline phase [20]. In this paper, the co-cracking of acetic acid and ethanol is investigated. The influence of reaction temperature and pressure on gasoline selectivity and quality is discussed in detail.
2 EXPERIMENTAL
HZSM-5 (Si/Al=25) zeolite catalyst was activatedat 550 °C for 6 h and sieved to 0.25-0.42 mm (40-60 mesh) before reaction. Catalytic experiments were carried out in a fixed-bed reactor. The reactor was a stainless steel tube with an inner diameter of 8 mm. About 2 g of catalyst was supported on quartz wool in the reactor. The reactant was a mixture of 30% acetic acid and 70% ethanol. The reactant was pumped by a HPLC pump and vaporized by a vaporizer, then entered the reactor using N2as a carrier gas. The pressure was maintained by N2, with a stabilized flow rate of 30 ml·min−1. The mass hourly space velocity (MHSV) of the reactants was held at 3 h−1. The reaction products were cooled by a condenser to separate the liquid products and non-condensable gas. Each experimental run lasted for 3 h. The conditions of the experiments are shown in Table 1.

Table 1 Experimental conditions
Both the gaseous and liquid products were analyzed. The gaseous products were quantified by an on-line gas chromatograph (Agilent 7890A). Light alkanes and olefins were separated on an HP-Plot Q capillary column with a flame ionization detector (FID). CO and CO2were separated on Porapak N, Porapak Q, and Carbon Sieve-11 columns and detected by a thermal conductivity detector. The GC oven was heated at 50 °C for 1 min, then the temperature was increased to 180 °C at 10 °C·min−1and held at this temperature. The liquid products obtained consisted of an easily separable crude gasoline phase and an aqueous phase. The crude gasoline phase was analyzed by a gas chromatography mass spectrometry system (Trace DSQ 2 system, manufactured by Thermal Fisher Company) with a 30 m×0.25 mm×0.25 μm Agilent DB-WAX capillary column. The GC oven was heated at 40 °C for 1 min, then the temperature was increased to 240 °C at 8 °C·min−1and held at this temperature. Data were acquired with Xcalibur software and with reference to the National Institute of Standards and Technology (NIST) mass spectral library database. Identified compounds were further quantified by the area normalization method. The residual reactants in both the crude gasoline phase and the aqueous phase were quantified by gas chromatography using the external reference method, thereby allowing calculation of the conversion of the reactants based on the feed mass.
The conversion of the reactants (Xi) and the selectivity for the liquid components (Si) are defined as follows.

where m represents the mass of the corresponding substance. The mass amounts of acetic acid (HAc) and ethanol (EtOH) were calculated by gas chromatography using the external reference method by assuming an ideal instrument condition. In the calculation of selectivity, the unconverted reactants were excluded from the received liquid products.
3 RESULTS AND DISCUSSION
3.1 Conversion rate of reactant
The conversion of acetic acid and ethanol at 2 MPa and different reaction temperatures is shown in Fig. 1 (a). The conversion of acetic acid and ethanol was above 99% at temperatures above 400 °C. However, the conversion of acetic acid declined sharply at temperatures below 400 °C, with 50.4% at 370 °C and 11.9% at 340 °C. Gayubo et al. [11] also found that the reactivity of acetic acid was very low below 400 °C during acetic acid cracking. Thus the cracking of acetic acid is seriously affected by reaction temperature. Temperatures above 400 °C could effectively activate acetic acid molecules and enhance the reactions, such as deoxidation, aromatization and alkylation, to produce liquid hydrocarbons.
Figure 1 (b) displays the conversion of acetic acid and ethanol at 400 °C and different pressures. Ethanol showed a conversion rate of 97% at atmospheric pressure and higher pressures. Acetic acid was also almost completely converted under pressurized conditions while its conversion rate was only 81.9% at atmospheric pressure. Therefore, pressurized conditions are beneficial for the conversion of acetic acid.
3.2 Selectivity of liquid product

Figure 1 Conversion of reactants under different conditionsHAc;EtOH

Figure 2 Selectivitys of liquid products under different conditionsaqueous phase;crude gasoline phase
Both aqueous phase and crude gasoline phase were obtained from the co-cracking of acetic acid and ethanol. The upper crude gasoline phase layer was light yellow, while the bottom aqueous phase layer was quite clear. The relative quantities of aqueous phase and crude gasoline phase at 2 MPa and different temperatures are illustrated in Fig. 2 (a). At temperatures above 370 °C, the selectivity for the crude gasoline phase was constant at around 38%, and decreased to 14.0% at 340 °C because the conversion of acetic acid was very low. The activity of the catalyst was also lower at lower temperatures. Further aromatization and alkylation of light olefins were suppressed by the low catalyst activity.
The selectivity of liquid products at 400 °C under different pressures is shown in Fig. 2 (b). The selectivity of crude gasoline was only 5.8% under atmospheric pressure, and it was 42.8%, 39.0% and 35.8% at 1, 2, and 3 MPa, respectively. This demonstrated that at a constant temperature of 400 °C, increasing reaction pressure from 0.1 MPa to 1 MPa favored the formation of crude gasoline obviously.
3.3 Composition of the crude gasoline phase
GC-MS analysis was used for the qualitative and quantitative analysis of the crude gasoline phase. Fig. 3 presents the main components in the crude gasoline phases obtained at 1 and 2 MPa with the temperature maintained at 400 °C, which were mainly aliphatic and aromatic hydrocarbons. Their carbon numbers range from 7 to 10, including toluene, dimethylbenzene and methyl-ethyl benzene, which are important components in the commercial gasoline.
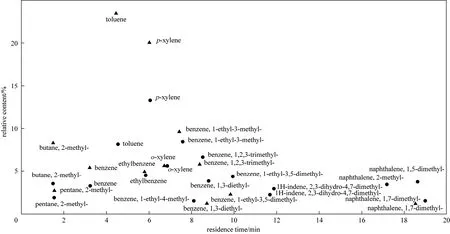
Figure 3 Main components in the crude gasoline phase obtained at 400 °C● 1 MPa; ▲ 2 MPa
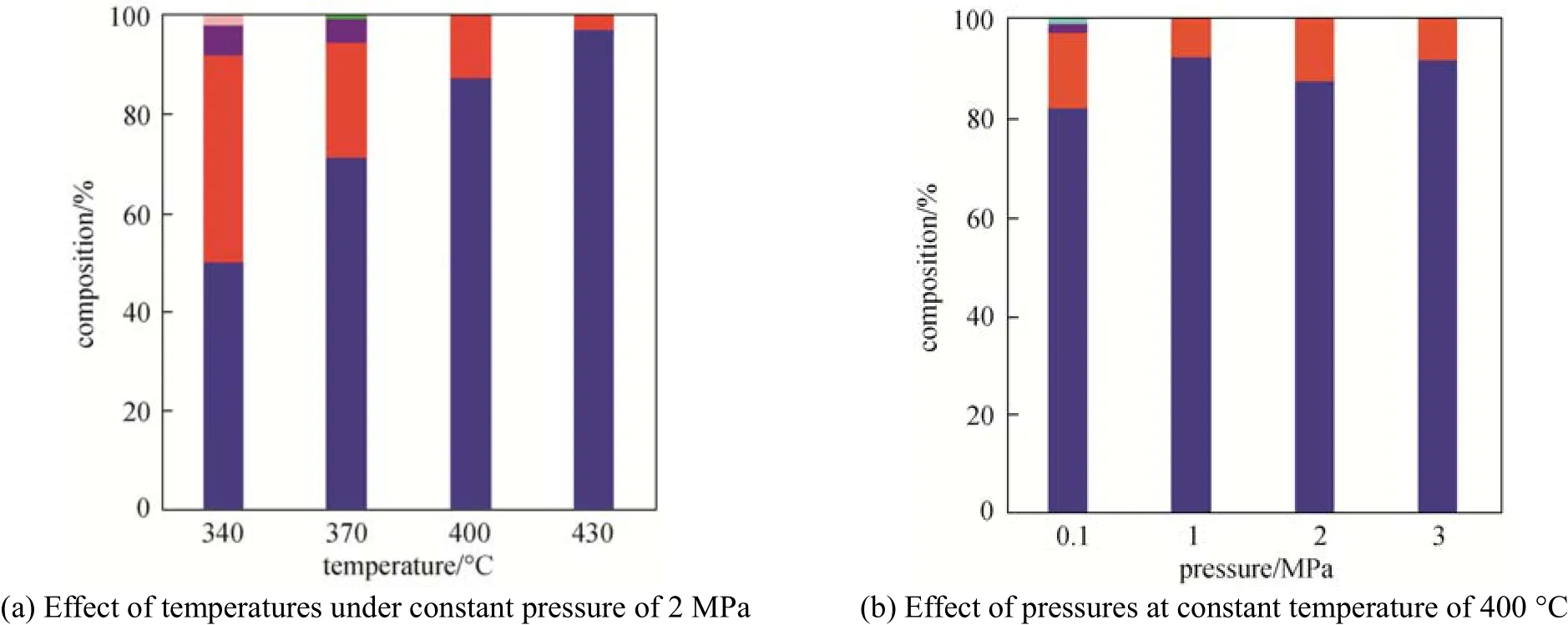
Figure 4 Composition of the crude gasoline phase from different conditionsaromatics;aliphatics;alcohols;esters;ketones;phenols
To make a clear comparison of the quality of the crude gasoline phases from different conditions, the compounds in the crude gasoline phase are classified into aromatics, aliphatics, alcohols, esters, ketones and phenols, and the results are summarized in Fig. 4. The crude gasoline phases produced at 400 °C and 430 °C under 2 MPa were pure hydrocarbons, with 87.7% and 97.5% of aromatic hydrocarbons, respectively. The content of oxygenated compounds in the crude gasoline phase was increased using cracking temperatures below 400 °C, giving an inferior quality crude gasoline phase. For example, the gasoline phase at 340 °C and 2 MPa had 1.5% of ethyl acetate and 5.8% of 1-pentanol. Because of the molecular structure of these oxygenated by-products, alcohol by-products were probably generated from the incomplete deoxygenation of ethanol and its intermediates with hydroxyl, while esters formed from the esterification reaction between ethanol and acetic acid. The deoxygenation process was incomplete as the low temperature reduced the activation of the reactant and the effectiveness of the catalyst. Consequently, temperatures above 400 °C must be used in order to achieve complete deoxygenation. It is noticed that the content of aromatics in the gasoline phase increases with temperature, which means that the aromatization reaction depends on the cracking temperature. Other researchers have observed similar phenomena in the catalytic cracking of model bio-oil compounds [11].
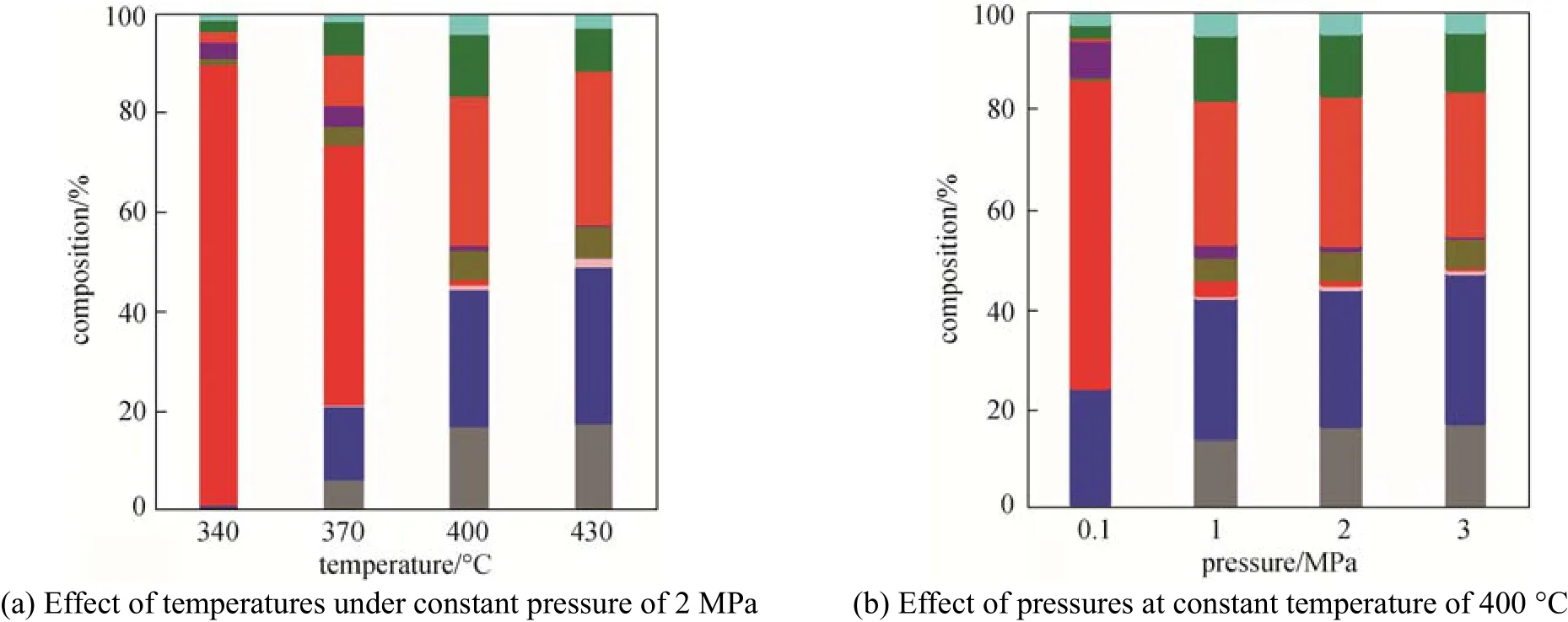
Figure 5 Composition of the outlet gas from different conditionsCO;CO2;CH4;C2H4;C2H6;C3H6;C3H8;C4H8;C4H10
The composition of crude gasoline at 400 °C and different pressures is shown in Fig. 4 (b). The crude gasoline phase from 1, 2 and 3 MPa is of high quality. The total proportions of hydrocarbons were 100%, and the proportions of the corresponding aromatics reached 93.1%, 87.7% and 92.0%, respectively. Although the proportion of hydrocarbons in crude gasoline phase under 0.1 MPa was 97%, the selectivity of gasoline phase was as low as 5.8%. Therefore, the catalyst deactivation may be the main reason for the decline in the selectivity of the crude gasoline phase because the olefins are released before they are aromatized to form liquid hydrocarbons, as a result of catalyst deactivation. The deposition of larger aromatic molecules is considered to be the major reason for catalyst deactivation. Nevertheless, since the inert gas N2remains at a certain pressure in the reactor, the partial pressure and diffusion velocity of reactants and intermediate products are reduced, avoiding the destruction of catalyst structure to some extent.
According to the above results, co-cracking of acetic acid and ethanol under pressurized condition and at temperatures above 400 °C benefits the conversion of reactants, giving high selectivity and high quality crude gasoline.
3.4 Composition of the outlet gas
Figure 5 (a) shows the composition of the outlet gas at 2 MPa. At 400 °C and 430 °C, the concentrations of COxin the gas effluent were around 45%, because the complete deoxygenation took place through decarbonyl and decarboxyl reactions at low effective H/C of acetic acid. The proportion of C3H8was also as high as 30%. As a saturated hydrocarbon, C3H8possibly came from the aromatization of intermediate olefins. The concentration of COxin the gas effluent decreased as the temperature decreased, from 21.1% at 370 °C to 0 at 340 °C, which is consistent with the variation of the conversion of the reactants, while the concentration of C2H4was higher under these two conditions, reaching 88.2% at 340 °C. The reason may be that lower temperature restrains the effectiveness of catalyst, and these intermediate olefins are released before the aromatization reactions.
The compositions of the gas effluent at 400 °C and different pressures are illustrated in Fig. 5 (b). The main gas products under pressurized conditions were COxand C3H8, while the concentration of COxdecreased to 24.5% at atmospheric pressure. Meanwhile, the concentration of C2H4was as high as 61.9%, which indicates the low effectiveness of the catalyst.
3.5 Catalyst coking analysis
The mass loss rates of the spent catalysts were measured by TG to investigate the carbon deposition during the catalytic cracking process, as shown in Fig. 6. The mass loss rates of the catalysts at 400 °C/0.1 MPa and 340 °C/2 MPa were 27.4% and 24.3%, respectively, much higher than that at 400 °C/2 MPa, which was only 10.5%. Thus carbon deposition will occur more easily on the catalyst in the catalytic cracking at low temperature or atmospheric pressure, which may lead to catalyst deactivation and decrease the selectivity and quality of the crude gasoline phase.
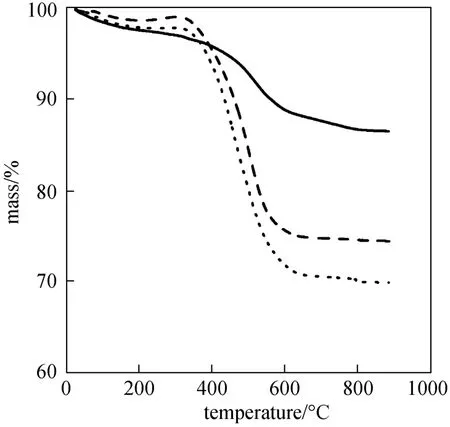
Figure 6 TG curves of catalysts for 30% acetic acid and 70% ethanol 400 °C/2 MPa; 340 °C/2 MPa; 400 °C/0.1 MPa
4 CONCLUSIONS
A study on the co-cracking of acetic acid and ethanol was carried out in a fixed bed reactor. The influence of reaction temperatures and pressures on the conversion of reactants, and the selectivity and composition of the crude gasoline were investigated. Temperatures below 400 °C limited the productivity of the catalyst, resulting in low conversion of acetic acid and incomplete deoxygenation of reactants. The selectivity of crude gasoline was as low as 5.8% under atmospheric pressure, because the catalyst was easily deactivated. The co-cracking of acetic acid and ethanol was very efficient at temperatures above 400 °C and the pressures above 1 MPa. The conversion of reactants was over 99% under 400 °C/1 MPa, and the crude gasoline had a selectivity of 42.79% with 100% liquid hydrocarbons.
REFERENCES
1 Butler, E., Devlin, G., Meier, D., McDonnell, K., “A review of recent laboratory research and commercial developments in fast pyrolysis and upgrading”, Renew. Sust. Energy Rev., 15 (8), 4171-4186 (2011).
2 Bridgwater, A.V., “Review of fast pyrolysis of biomass and product upgrading”, Biomass Bioenergy, 38, 68-94 (2012).
3 Czernik, S., Bridgwater, A.V., “Overview of applications of biomass fast pyrolysis oil”, Energy Fuels, 18 (2), 590-598 (2004).
4 Mortensen, P.M., Grunwaldta, J.D., Jensena, P.A., Knudsenc, K.G., Jensen, A.D., “A review of catalytic upgrading of bio-oil to engine fuels”, Appl. Catal. A, 407, 1-19 (2011).
5 Park, H.J., Jeon, J.K., Suh, D.J., Suh, Y.W., Heo, H.S., Park Y.K.,“Catalytic vapor cracking for improvement of bio-oil quality”, Catal. Surv. Asia, 15 (3), 161-180 (2011).
6 Valle, B., Gayubo, A.G., Aguayo A.T., Olazar, M., Bilbao, J., “Selective production of aromatics by crude bio-oil valorization with a nickel-modified HZSM-5 zeolite catalyst”, Energy Fuels, 24 (3), 2060-2070 (2010).
7 Luo, Z., Wang, S., Liao, Y., Zhou, J., Gu, Y., Cen, K., “Research on biomass fast pyrolysis for liquid fuel”, Biomass Bioenergy, 26 (5), 455-462 (2004).
8 Guo, X., Wang, S., Wang, Q., Guo, Z., Luo, Z., “Properties of bio-oil from fast pyrolysis of rice husk”, Chin. J. Chem. Eng., 19 (1), 116-121 (2011).
9 Adjaye, J.D., Bakhshi, N.N., “Production of hydrocarbons by catalytic upgrading of a fast pyrolysis bio-oil. Part I: conversion over various catalysts”, Fuel Process. Technol., 45 (3), 161-183 (1995).
10 Gayubo, A.G., Aguayo, A.T., Atutxa, A., Aguado, R., Bilbao, J.,“Transformation of oxygenate components of biomass pyrolysis oil on a HZSM-5 zeolite. I. Alcohols and phenols”, Ind. Eng. Chem. Res., 43 (11), 2610-2618 (2004).
11 Gayubo, A.G., Aguayo, A.T., Atutxa, A., Aguado R., Olazar, M., Bilbao, J., “Transformation of oxygenate components of biomass pyrolysis oil on a HZSM-5 zeolite. H. Aldehydes, ketones, and acids”, Ind. Eng. Chem. Res., 43 (11), 2619-2626 (2004).
12 Mentzel, U.V., Holm, M.S., “Utilization of biomass: conversion of model compounds to hydrocarbons over zeolite H-ZSM-5”, Appl. Catal., A, 396, 59-67 (2011).
13 Lin, Y., Tanaka, S., “Ethanol fermentation from biomass resources: current state and prospects”, Appl. Microbiol. Biotechnol., 69 (6), 627-642 (2006).
14 Wang, S.R., Guo, X., Liang, T., Zhou, Y., Luo, Z., “Mechanism research on cellulose pyrolysis by Py-GC/MS and subsequent density functional theory studies”, Bioresour. Technol., 104, 722-728 (2012).
15 Peng, Y., Wu, S., “The structural and thermal characteristics of wheat straw hemicelluloses”, J. Anal. Appl. Pyrolysis, 88 (2), 134-139 (2010).
16 Demirbas, A., “The influence of temperature on the yields of compounds existing in bio-oils obtained from biomass samples via pyrolysis”, Fuel Process. Technol., 88 (6), 591-597 (2007).
17 Wang, S., Gu, Y., Liu, Q., Yao, Y., Guo, Z., Luo, Z., Cen, K., “Separation of bio-oil by molecular distillation”, Fuel Process. Technol., 90 (5), 738-745 (2009).
18 Guo, Z., Wang, S., Gu, Y., Xu, G., Li, X., Luo, Z., “Separation characteristics of biomass pyrolysis oil in molecular distillation”, Sep. Purif. Technol., 76 (1), 52-57 (2010).
19 Guo, Z., Wang, S., Zhu, Y., Luo, Z., Cen, K., “Separation of acid compounds for refining biomass pyrolysis oil”, J. Fuel Chem. Technol., 37 (1), 49-52 (2009).
20 Wang, S., Cai, Q., Guo, Z., Wang, Y., Wang, X., “Renewable gasoline produced by co-cracking of methanol and ketones in bio-oil”, Bioresourc., 7 (4), 5019-5031 (2012).
10.1016/S1004-9541(14)60013-6
2012-10-20, accepted 2013-01-30.
* Supported by the National Natural Science Foundation of China (51276166), the National Science Technology Supporting Plan Through Contract (2011BAD22B06), the Zhejiang Provincial Natural Science Foundation (R1110089) and the Program for New Century Excellent Talents in University (NCET-10-0741).
** To whom correspondence should be addressed. E-mail: srwang@zju.edu.cn
 Chinese Journal of Chemical Engineering2014年1期
Chinese Journal of Chemical Engineering2014年1期
- Chinese Journal of Chemical Engineering的其它文章
- Influence of Design Margin on Operation Optimization and Control Performance of Chemical Processes*
- Measurement and Modeling for the Solubility of Hydrogen Sulfide in Primene JM-T*
- Enhancing Structural Stability and Pervaporation Performance of Composite Membranes by Coating Gelatin onto Hydrophilically Modified Support Layer*
- Photocatalytical Inactivation of Enterococcus faecalis from Water Using Functional Materials Based on Natural Zeolite and Titanium Dioxide*
- A Group Contribution Method for the Correlation of Static Dielectric Constant of Ionic Liquids*
- Interaction Analysis and Decomposition Principle for Control Structure Design of Large-scale Systems*