
LiH和LiT与H2O反应机理研究
2014-08-08 02:50苟振志罗忠辉喻凤梅
原子能科学技术 2014年12期
苟振志,何 彬,罗忠辉,喻凤梅
(1.第二炮兵工程大学,陕西 西安 710025;2.中国人民解放军96421部队,陕西 宝鸡 721012)
LiT和LiD一样,是一种重要的核工业原料,也是武器级核材料重要的原料,但在长期贮存过程中,该部件容易与空气中的水分结合而使得品位降低,达不到武器级部件标准的要求而退役。文献[1]研究了LiD与H2O的反应机理,结果表明,LiD与H2O的反应存在两个通道,第1通道的反应速率大于第2通道的反应速率。本文拟用二阶微扰理论和Gaussian03[2-7]软件包对LiT和LiH与H2O的反应进行研究。
1 计算方法
LiT、LiH与LiD一样,在与H2O的反应中是氚原子取代LiH中的氢原子。理论上推测,LiT与H2O的反应具有两个通道:

计算发现,第2个反应通道不存在中间体和过渡态,所以LiT与H2O反应路径只有1个。
本文采用Gaussian03软件包中的量子化学从头计算法[8-9],在6-311G(d)水平上,用量子化学MP2方法计算LiH和LiT在H2O气氛中腐蚀反应各驻点的分子几何结构,通过频率分析确定反应中间体和过渡态,同时对内禀反应坐标(IRC)进行计算,确定反应过渡态的真实性。经过优化后在CCSD/6-311++G(d,p)水平上计算其能量,并比较各物质间的相对能,这样既提高了精度又节省了时间。此外,CCSD是在HF基础上发展出的考虑电子相关的耦合簇方法,因HF方法具有局限性,虽然它为许多体系的计算提供了一个合理的模型,但未能充分考虑电子相关,特别是瞬时的电子相关作用,所以对这类作用很明显的体系,HF方法不能给出很好的描述。两反应的反应速率常数K由下式确定:
K=Ae-Ea/RT
(1)
式中:Ea为反应活化能,其值为过渡态与中间体或反应物能量(经零点能校正)的差值;R为普适气体常数;T为热力学温度;A为指前因子,由式(2)计算:
(2)

2 结果与讨论
2.1 反应历程及其能量分析
表1为基于MP2/6-311G(d) 二阶微扰理论方法优化得到的反应物R、中间体M、过渡态TS和产物P等驻点的几何构型参数。表2为反应体系各驻点的零点能ZPE、包含零点能的MP2/6-311G(d)的总能量E、CCSD/6-311++G(d,p)的计算能量及以CCSD-ZPE水平上反应物能量为参照的相对能量Erel。LiH和LiT与H2O反应的能级变化示于图1。由图1可知,2个反应中,产物的能量均低于反应物的,即均为放热反应。各反应通道的势垒ΔE及室温(298 K)下的焓变ΔH列于表3,产物的能量较反应物的能量低40 kJ/mol以上,从热力学角度来说均是可能发生的反应。
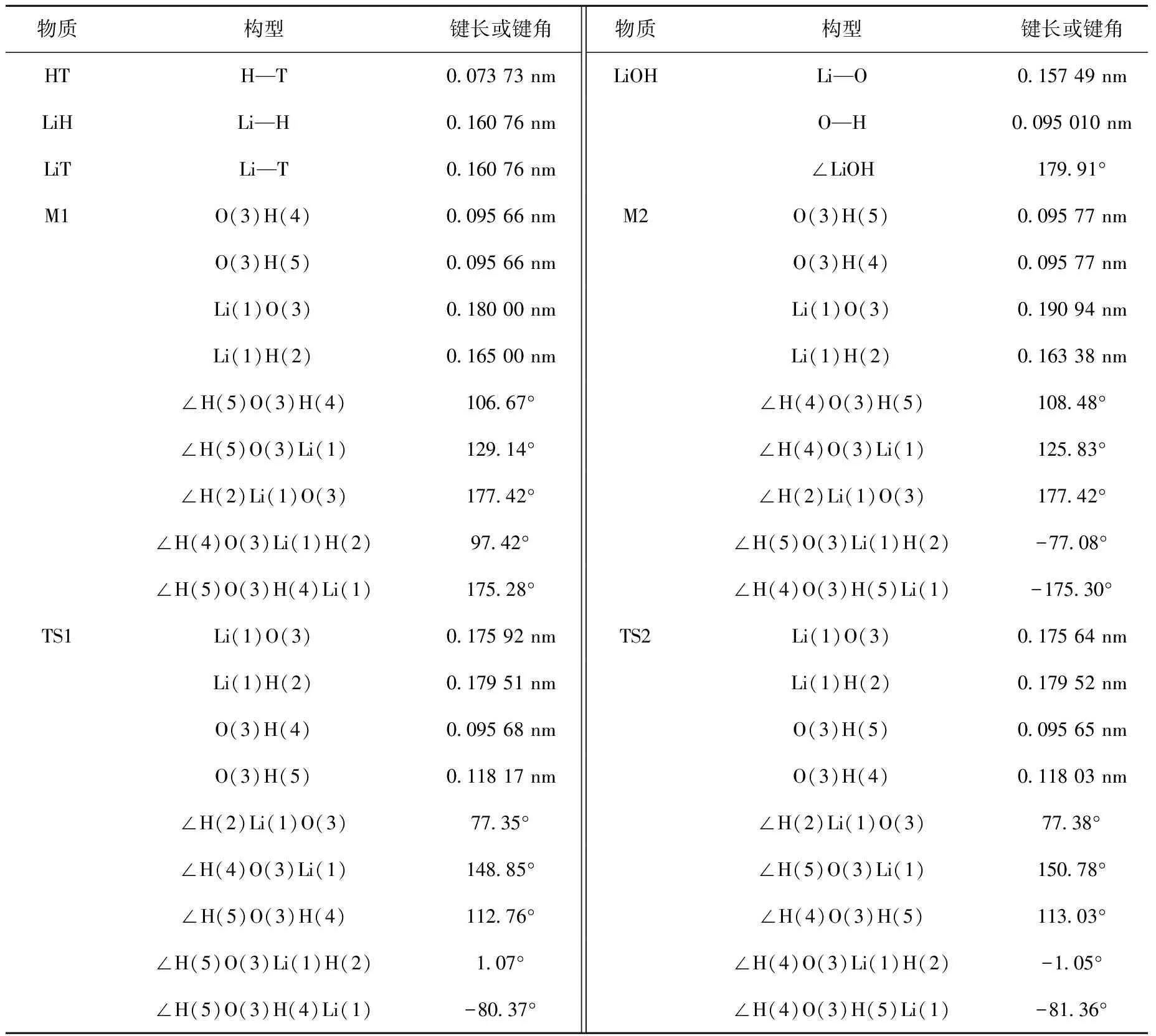
表1 LiH和LiT与H2O反应的各中间体、过渡态和产物及反应物的几何构型参数

表2 LiH和LiT与H2O反应路径各驻点的E、零点能ZPE、CCSD、Erel及势垒ΔE

图1 LiH和LiT与H2O反应的相对能量示意图

表3 LiH和LiT与H2O反应中各反应通道的势垒ΔE及焓变ΔH

图2 LiH+H2OLiOH+H2的反应历程
2.2 振动频率分析
通过振动并结合分子结构和反应机理分析了两个反应的反应物、中间体和过渡态以及生成物的频率,结果列于表4。由表4可知,反应物、中间体和生成物的频率均为正值,说明它们为势能面上的稳定点。两个过渡态均只有1个虚频,通过对优化得到的过渡态进行内禀反应坐标法计算,结果表明它们是反应途径上的真实过渡态。
2.3 反应热力学和动力学参数的计算
根据式(1)、(2)计算的LiH和LiT与H2O反应的焓变ΔHm及反应速率常数列于表5,计算条件为压力p=100 kPa,T=298.15 K。表5数据表明,两反应均为放热反应,且反应速率常数较大,反应活化能低,因此两反应均很容易实现。

表4 LiH和LiT与H2O反应各驻点的振动频率

表5 LiH和LiT与H2O反应的相关热力学和动力学参数
3 结论
本文用从头计算的MP2方法,利用Gaussian03软件分别对LiH和LiT与H2O的反应机理进行了理论研究。优化在CCSD/6-311++G(d,p)水平上计算的能量,并比较了各物质间的相对能,这样既提高了精度又节省了时间。优化了两反应的中间体和过渡态,并通过内禀反应坐标法确定了过渡态位于正确的反应路径上。经过大量的过渡态搜索工作,从理论上得到LiH和LiT与H2O反应均仅有1条反应通道的结论。LiH与H2O反应活化能和速率常数分别为8.95 kJ/mol和3.75×1010(mol·dm-3)-1/s,LiT与H2O反应活化能和速率常数分别为9.92 kJ/mol和1.72×1010(mol·dm-3)-1/s。
参考文献:
[1] 罗忠辉,何彬,牛莉博,等. LiD与H2O反应机理及动力学[J]. 原子能科学与技术,2010,44(1):80-84.
LUO Zhonghui, HE Bin, NIU Libo, et al. Theoretical study on mechanism and kinetics for reaction of LiD with H2O[J]. Atomic Energy Science and Technology, 2010, 44(1): 80-84(in Chinese).
[2] RAGHAVACHARI K, POPLE J A, REPLOGLE E S, et al. Fifth-order moller-plesset perturbation theory: Comparison of existing correlation methods and implementation of new methods correct to fifth-order[J]. J Phys Chem, 1990, 94: 5 579-5 585.
[3] HOHENBERG P, KOHN W. Inhomogeneous electron gas[J]. Phys Rev B, 1964, 136: 864-871.
[4] KOHN W, SHAM L J. Self-consistent equations including exchange and correlationeffects[J]. Phys Rev A, 1965, 140: 1 133-1 138.
[5] SLATE J C. Quantum theory of molecular and solids, Vol. 4: The self-consistent field for molecular and solids[M]. New York: McGraw-Hill, 1974.
[6] SALAHUB D R, ZERNER M C. The challenge of d and f electrons[M]. Washington D. C.: ACS, 1989.
[7] PARR R G, YANG W. Density-functional theory of atoms and molecules[M]. Oxford: Oxford University Press, 1989.
[8] 李来才,周红平,田安民. F原子与臭氧反应机理的量子化学研究[J]. 物理化学学报,2002,18(1):59-61.
LI Laicai, ZHOU Hongping, TIAN Anmin. Quantum chemical study on the reaction mechanism of ozone with fluorine atoms[J]. Acta Physico-chimica Sinica, 2002, 18(1): 59-61(in Chinese).
[9] 李宝宗. 甲醛与甲酰胺相互作用的从头算研究[J]. 化学物理学报,2004,17(4):433-436.
LI Baozong. Ab initio study on the interaction between formaldehyde and formamide[J]. Chinese Journal of Chemical Physics, 2004, 17(4): 433-436(in Chinese).
[10] 何惊华. 金属锂及氢化锂在水气氛中反应机理的理论研究[D]. 成都:四川师范大学,2005.
猜你喜欢
北京航空航天大学学报(2022年5期)2022-06-06
中国园林(2018年7期)2018-08-07
电脑知识与技术(2018年3期)2018-03-21
中学化学(2017年5期)2017-07-07
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
中学化学(2016年4期)2016-05-30
党的生活·党员电教与远程教育(2016年3期)2016-02-26
源流(2015年8期)2015-09-16
学习月刊(2015年2期)2015-07-09
