
高效液相色谱法测定利鲁唑片有关物质及含量
2014-11-08 08:36陈杰,李展
中国药业 2014年12期
陈 杰,李 展
(河南省食品药品检验所,河南 郑州 450003)
利鲁唑为谷氨酸盐拮抗剂,可抑制脑内神经递质的释放,对谷氨酸能神经突触传递有抑制作用,并抑制γ氨基丁酸(GABA)、多巴胺、谷氨酸的再摄取;还可明显抑制兴奋性氨基酸的活性;可稳定电压依赖性钠通道的失活状态,具有明显的神经保护作用。其主要用于运动神经元疾病的治疗。目前,国内共有3家企业生产利鲁唑片,其有关物质测定方法均为高效液相色谱(HPLC)法,色谱条件及测定方法有一定差异[1-3];含量测定方法均为紫外光光(UV)外标法。笔者参照USP33[4],以原研产品为基础,征集这3家企业共9批样品,在进行质量研究并比对质量的基础上,建立有关物质及含量测定的HPLC测定方法。
1 仪器与试药
Agilent 1200型高效液相色谱仪(美国Waters公司);AX205型电子天平(Metler公司)。利鲁唑对照品(含量99.1%,鲁南制药股份有限公司);力如太(规格为每片50 mg,AventisPharmas公司,批号为H112405);乙腈为色谱纯。
2 方法与结果
2.1 有关物质测定
2.1.1 色谱条件
色谱柱:用十八烷基硅烷键合硅胶为填充剂,Agilent TC-C18柱(250 mm ×4.6 mm,5 μm);流动相:乙腈 -水(9 ∶11);检测波长:221 nm;流速:1.0 mL/min。理论板数按利鲁唑峰计算不得低于2 000。
2.1.2 溶液制备
取本品研成细粉,精密称取细粉适量,加流动相制成每1 mL中约含500 μg的溶液,作为供试品溶液。精密量取供试品溶液1 mL,置100 mL容量瓶中,用流动相稀释至刻度,摇匀,作为对照溶液。
2.1.3 测定方法
取对照溶液10 μL,注入液相色谱仪,调节检测灵敏度,使主峰高约为满量程的20%,再精密量取供试品溶液和对照溶液各10 μL,分别注入液相色谱仪,记录色谱图至供试品溶液主峰的保留时间的3倍。
2.1.4 方法学考察
专属性试验:取本品适量,分别进行酸破坏(5 mol/L HCl)、碱破坏(0.5 mol/L NaOH)、氧化破坏(2%H2O2)、热破坏(100 ℃水浴破坏)和光破坏(照度3 000 lx,7 d)试验。结果见图1。表明拟订的色谱条件能有效分离破坏后降解产物。
检出限确定:取对照溶液,逐级稀释,按拟订的色谱条件进行测定。结果检出限为0.32 ng。
稳定性试验:取同一供试品溶液,分别于 0,2,4,8,12 h时依法测定,有关物质检测结果基本一致,表明用流动相稀释制备的供试品溶液稳定性良好。
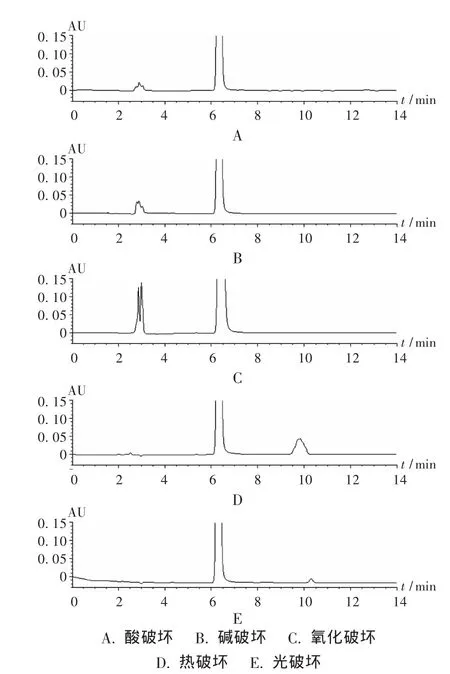
图1 专属性实验色谱图
2.1.5 样品检测
取征集的各企业产品及原研产品,按拟订的方法检测。各企业产品原有关物质检测方法见表1,检测结果见表2。

表1 各厂家利鲁唑片有关物质测定方法比较
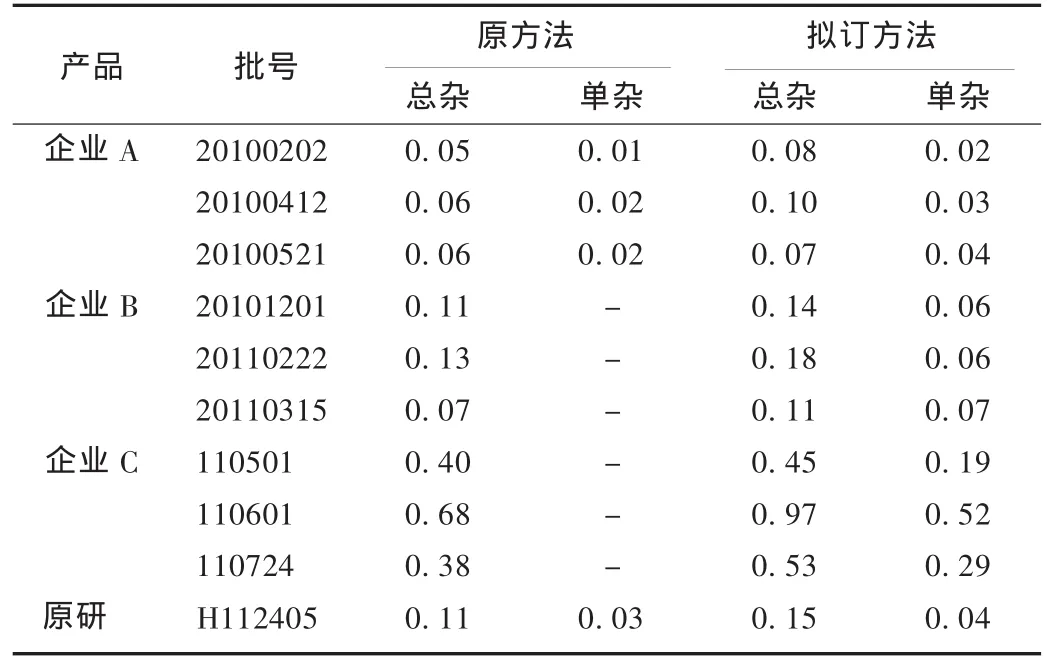
表2 各企业产品有关物质测定结果(%)
2.2 含量测定
2.2.1 色谱条件
色谱柱及流动相:同有关物质测定方法;检测波长:254 nm;流速:1.0 mL/min;进样量:10 μL。理论板数按利鲁唑峰计算不得低于2 000。
2.2.2 溶液制备
取本品研成细粉,精密称取细粉适量,加流动相制成每1 mL中约含50 μg的溶液,作为供试品溶液。取利鲁唑对照品适量,同法制成相同质量浓度的对照品溶液。
2.2.3 方法学考察
加样回收试验:分别取利鲁唑对照品8,10,12 mg各3份,分别加适量的辅料(磷酸氢钙、微晶纤维素、微粉硅胶、硬脂酸镁、交联聚维酮K30、羧甲淀粉钠),依法制成供试品溶液,测定含量,计算回收率。结果平均回收率为99.98%,RSD=0.35%(n=9)。
精密度试验:取回收试验中由10 mg利鲁唑对照品溶液制备的同一份供试品溶液,连续进样6次。结果峰面积的 RSD=0.41%(n=6)。
线性关系考察:取利鲁唑对照品 5,8,10,12,15 mg,依质量法制成对照品溶液并测定,以利鲁唑质量浓度为横坐标、峰面积为纵坐标进行线性回归,回归方程为 Y=28 439 X-796.55,R2=0.9998(n=5)。结果表明,利鲁唑进样量线性范围为0.25 ~0.75μg。
2.2.4 样品含量测定
取各企业产品及原研产品,按拟订方法进样测定,并与采用UV法测定结果比较。结果见表3。
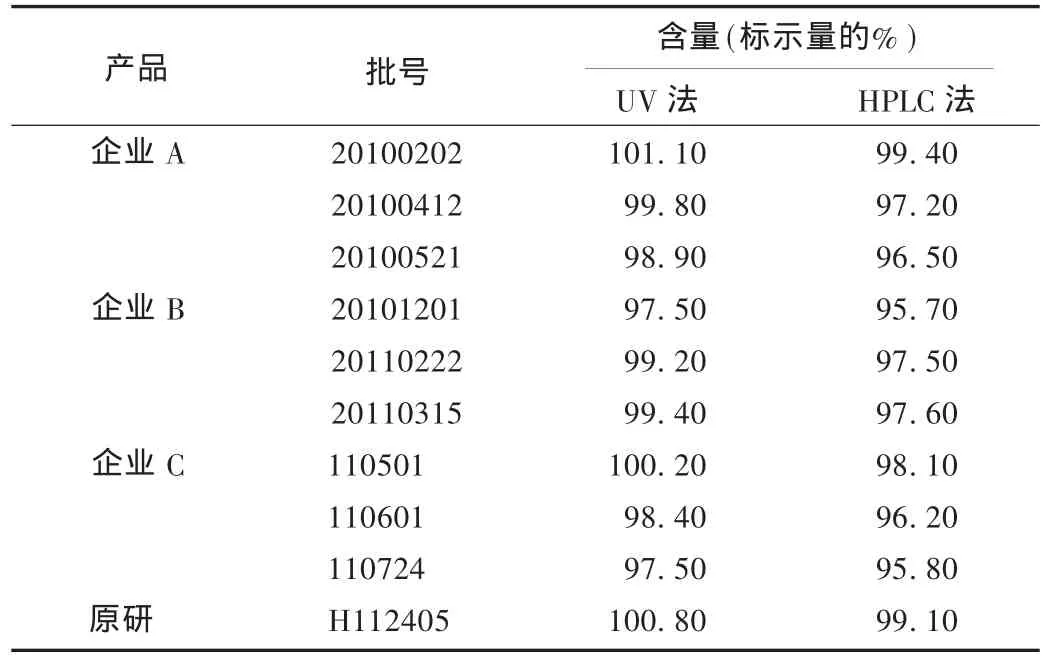
表3 各企业产品利鲁唑含量测定结果
3 讨论
国内3家生产利鲁唑原料的企业,生产工艺各不相同,产品晶型和杂质各不相同。本试验中统一了利鲁唑片的有关物质测定方法,经试验比对,与其他3种色谱体系相比,拟订方法对杂质的分离度更好,与原方法相比增加了单杂的控制。由于检测波长改为221nm,加大了供试品溶液质量浓度,因此杂质峰检出更多,总杂检出值更大。
与原方法相比,含量测定改为HPLC法后含量测定结果下降约2%,分析原因可能是原方法受到来自辅料及杂质的干扰所致。HPLC法测定含量,不仅提高了方法专属性,且排除了原料中杂质的干扰。
[1]YBH13482004,国家药品标准[S].
[2]YBH17352004,国家药品标准[S].
[3]YBH16452005,国家药品标准[S].
[4]The Vnited states Pharmacopeial convention.U.S.Pharmacopeial 33-NF28[S].Washington:The Broad of Trusfees,2010:2 124-2 125.
猜你喜欢
四川蚕业(2022年2期)2022-11-19
色谱(2022年11期)2022-11-10
色谱(2022年10期)2022-10-13
山东冶金(2022年3期)2022-07-19
色谱(2022年7期)2022-06-25
玩具世界(2022年1期)2022-06-05
色谱(2022年4期)2022-04-01
河南农业·综合版(2022年2期)2022-03-18
环境保护与循环经济(2021年7期)2021-11-02
山西中医药大学学报(2020年2期)2020-06-06
