
苯并噻唑2-甲酸的合成及应用研究进展
2014-12-23 01:01侯巍巍白金泉吴翠敏
应用化工 2014年8期
侯巍巍,白金泉,吴翠敏
(北京工业大学 环境与能源工程学院 化学化工系,北京 100124)
杂环化合物对改善生命体机能有很大的作用,其与材料科学有着十分密切的关系。目前,对杂环化合物的研究已越来越深入,并不断改善、影响着人们的生活。其中,苯并噻唑杂环类化合物是有机合成反应的先导骨架与母体之一。自1879 年Hofmann 首次介绍了2-氯和2-苯基苯并噻唑以来,越来越多的衍生物被合成。该类化合物因具有药理及生物活性而被广泛应用于农药、医药等领域[1-3]。
苯并噻唑衍生物的取代分为苯环上和2 位上的取代,而2 位取代衍生物活性基团对其本身活性影响较大。李炎等[4]则介绍了一系列2-取代苯并噻唑衍生物的合成方法。本文将对苯并噻唑2-甲酸的合成及应用进展作简要综述。
1 合成方法
1.1 CO2 羧化法
二氧化碳(CO2)被视为廉价、可再生、来源丰富且用途广泛的能源。在过去的十几年内,其作为对过渡金属介导激活剂已受到越来越多的关注。CO2作为C1 源和与它羧化的亲核物质间可以形成新的C—C 骨架,被视为羧酸及其衍生物合成的极具优势的原料。但是由于CO2极高的热力学及动力学稳定性,使亲核试剂被限制在某些具有金属活性的不饱和烃类及活性金属有机试剂如格氏试剂。为了探索CO2对亲核试剂的羧化反应,许多科研工作者做了不同程度的研究。
1.1.1 CO2直接羧化法 Oleg Vechorkin 等[5]用CO2羧化法,通过改变催化剂的种类等条件合成了苯并噻唑2-甲酸。

图1 直接羧化苯并噻唑制备苯并噻唑2-甲酸Fig.1 Direct carboxylation of benzothiazolea

表1 直接羧化苯并噻唑的反应条件Table 1 Conditions for direct carboxylation of benzothiazolea
苯并噻唑2-甲酸(见图1),具体方案及转化率如表1 所示。由表1 实验数据可知,该课题组在没有金属催化剂只在Cs2CO3基质的条件中,用CO2作C1 源合成了苯并噻唑2-甲酸,转化率达到100%,反应式见图2,反应得到的杂环酸与酯在医药及材料科学中具有重要的应用价值。与其他方法相比,此方法具有原子经济性的优势,适用于工业生产。

图2 直接羧化苯并噻唑制备苯并噻唑2-甲酸Fig.2 Direct carboxylation of benzothiazolea
1.1.2 卡宾金(Ⅰ)催化CO2羧化法 Ine I F Boogaerts 等[6]用(IPr)AuOH(IPr =1,3-二(二异丙基)苯并咪唑-2-自由基)作催化剂,用CO2作C1 源合成了苯并噻唑2-甲酸,产率为76%(图3)。该课题组的研究,为卡宾金属催化剂催化合成苯并噻唑2-甲酸的研究奠定了基础。
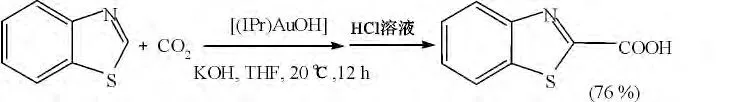
图3 用[(IPr)AuOH]羧化苯并噻唑Fig.3 Carboxylation of benzothiazole with[(IPr)AuOH]
1.1.3 卡宾铜(Ⅰ)催化CO2羧化法 Ine I F Boogaerts 等[7]用卡宾铜([Cu(IPr)OH](IPr =1,3-二(二异丙基)苯并咪唑-2-自由基))作催化剂,初步探索了CO2羧化苯并杂环化合物的机理,设想出催化羧化的机理环,并成功用苯并噻唑制备了苯并噻唑2-甲酸(图4)。目前,该课题组主要着力于分析催化剂成分不同对羧化机理产生的影响。

图4 用[Cu(IPr)OH]羧化苯并噻唑Fig.4 Carboxylation of benzothiazole with[Cu(IPr)OH]
Hiroshi Inomata 等[8]用1,2,3-三唑-5-甲基铜(Ⅰ)化合物([(TPr)CuCl])有效的催化CO2羧化苯并杂环化合物得到相应的酯,并将[(TPr)CuCl]与卡宾铜(Ⅰ)催化效果进行对比并用气相色谱检测产率,结果显示,[(TPr)CuCl]催化速率更快,产率更高;在反应3 h 后用[(TPr)CuCl]催化的反应产率已达到91%;反应5 h 后[(TPr)CuCl]和卡宾铜(Ⅰ)两种催化剂催化羧化的产率分别为93%和86%;反应8 h 后两者产率均趋于稳定。该课题组在此基础上,用[(TPr)CuCl]作催化剂,催化羧化了苯并噻唑类化合物(图5),用气相色谱法检测其产率可达到75%。此外,该课题组分析推测了[(TPr)CuCl]催化羧化的机理,并推测[(TPr)-CuCl]和卡宾铜(Ⅰ)的催化羧化机理可能很相似:首先,(TPr)CuCl 和t-BuOK 发生金属置换反应得到(TPr)Cu(OBut),然后利用铜(Ⅰ)醇盐与苯并噻唑反应将苯并噻唑的C—H 骨架去区质子化得到(TPr)(苯并噻唑)Cu,将CO2引入到Cu—C 键间得到铜酸盐,最终再利用金属置换反应得到羧酸钾盐;在整个过程中铜(Ⅰ)醇盐是可循环再生利用的,如图6 所示。该课题组对卡宾金属催化合成机理的推测,势必是卡宾催化合成羧酸研究进展中较为重要的一步。

图5 用[(TPr)CuCl]羧化苯并噻唑Fig.5 Carboxylation of benzothiazole with[(TPr)CuCl]

图6 可能的反应机理Fig.6 Plausible reaction mechanism
1.2 水解法
1.2.1 酯水解生成酸 Kamps Jos J A G 等[9]用苯并噻唑2-甲酸乙酯为原料,以甲醇为溶剂,在室温下于NaOH 溶液体系中反应1 h,最终得到苯并噻唑2-甲酸,产率为92%(图7)。

图7 酯水解生成苯并噻唑2-甲酸Fig.7 Hydrolysis of ester for 2-benzothiazolecarboxylic acid
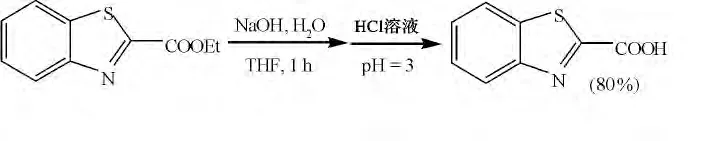
图8 酯水解生成苯并噻唑2-甲酸Fig.8 Hydrolysis of ester for 2-benzothiazolecarboxylic acid
在此基础上,采用同样的原理,通过改变反应液后处理的酸性条件,研究了制备苯并噻唑2-甲酸的方法:以苯并噻唑2-甲酸乙酯为原料,四氢呋喃(THF)作溶剂,在室温下于NaOH 和水的混合体系中反应1 h,并用HCl 调节溶液酸性到pH 值为3,最终得到苯并噻唑2-甲酸,产率为80%(图8)。
1.3 高锰酸钾氧化法
Vamsi Krishna K 等[10]用2-甲基苯并噻唑为原料,在酸性高锰酸钾(KMnO4)作氧化剂的条件下,合成了苯并噻唑2-甲酸。该课题组先将原料液处于KMnO4水溶液中回流,冷凝;再用H2SO4水溶液处理反应液,最终得到产物苯并噻唑2-甲酸(图9)。此制备方法,经过强酸催化与处理,对反应设备腐蚀较严重,并且最终反应液对环境危害性较大,因此,可能不太适宜工业化生产。

图9 苯并噻唑2-甲酸的合成Fig.9 Synthesis of 2-benzothiazolecarboxylic acid
1.4 Phillips 合成法
Walker Daniel P 等[11]用邻氨基苯硫酚与乙二酸二乙酯作原料,利用Phillips 合成法,回流4 h 后,在室温下,以NaOH 为助剂,甲醇为溶剂,反应1 h ,最终得到产品苯并噻唑2-甲酸(图10)。反应机理可能为:氮原子对羰基碳发生亲核进攻,得到酰胺中间体,随后硫原子又发生亲核进攻而脱水闭环。该方法是利用邻氨基苯硫酚合成苯并噻唑类衍生物常用的制备方法。

图10 苯并噻唑2-甲酸的合成Fig.10 Synthesis of 2-benzothiazolecarboxylic acid
1.5 其他合成方法
Ahmad M Farag 等在新的取代噻吩合成过程中得到了苯并噻唑2-甲酸;当用浓盐酸酸化噻吩衍生物反应液并经抽滤、水洗及干燥后,母液中出现了一种沉淀物,将沉淀物重结晶后用红外光谱等分析,结果显示,沉淀物为苯并噻唑2-甲酸。
此外,用二聚酯(见图11)在NaOH 溶液中,30 ℃条件下反应2 h,再经酸化最终也制得了苯并噻唑2-甲酸,如图11 所示。

图11 苯并噻唑2-甲酸的合成Fig.11 Synthesis of 2-benzothiazolecarboxylic acid
2 应用
2.1 医学及医药领域
2-取代苯并噻唑类衍生物作为一种含氮杂环化合物,具有广泛的生物活性与药理活性[12-14],被作为重要的药物合成结构单元。苯并噻唑2-甲酸,作为2-取代苯并噻唑化合物,是一种重要的有机医药中间体[15-18],在抗癌、消炎、抗病毒等方面发挥着重要的作用。
2.1.1 抗病毒作用严重急性呼吸系统综合症(severe acute respiratory syndrome,SARS)是一种具有高度传染性和致命性的呼吸道疾病,8 000 多人曾被感染;其中10%的感染者死在SARS 出现的几个月内。权威专家和世界卫生组织(WHO)之间合作研究出了新型冠状病毒(CoV),并把它作为快速识别SARS 感染病因的试剂。
Pillaiyar Thanigaimalai 等[19]介绍了低分子量肽SARS 冠状病毒胰凝乳蛋白酶(3CL)抑制剂的设计与合成方法。3CL 也称为主蛋白酶,在形成病毒聚蛋白的过程以及控制复制酶活性中起着很大的作用。一系列的研究结果显示,二肽型蛋白酶抑制剂对3CL 有中度至良好的抑制活性。特别引起注意的是化合物A 和B(图12)的抑制活性尤其好,其抑制常数(Ki)值分别是0.39,0.33 mmol/L。该课题组模拟了化合物A 与SARS 冠状病毒3CL 蛋白酶的结合作用。通过对肽类化合物以及其对SAR 有效的抑制活性的初步研究,得知化合物的某些结构特征增强了其自身的抑制活性。这些结构分别是:苯并噻唑的头部处于S1’位,一个-内酰胺单元处于S1位,适当疏水性的亮氨酸处于S2 位,处于N-芳基甘氨酸与骨干H 供体间一个H 处于S3 位。研究结果表明作为有效部位,苯并噻唑是P1’部位最合适的结构单元。在此研究基础上,该课题组以苯并噻唑2-羧酸衍生物为母体(见图13),改变连接基团的结构,设计合成了一系列具有新的P3 骨架的二肽蛋白酶抑制剂,可以有效的抑制SARS-CoV 3CLpro的活性[20]。此研究为抑制SARS 感染做出了巨大的贡献。

图12 化合物A 与B Fig.12 Compound A and B

图13 苯并噻唑2-羧酸衍生物Fig.13 Derivatives of 2-benzothiazolecarboxylic acid
2.1.2 消炎作用苯并噻唑2-甲酸不仅可以用来制备抵抗病毒的药物,还可以用于合成抗炎症性疾病的药物[21]。趋化因子受体(chemokine receptors,CCR1),是一类特异性地吸引白细胞的小分子蛋白质,通过与配体的相互作用,调解了白细胞的活化和迁移,对炎性疾病的发展至关重要,其结构如图14所示。

图14 趋化因子受体Fig.14 Chemokine receptors
Cullen L 等[22]用迭代并行适度撞击的方法合成了一系列新的,有效的CCR1 抑制剂。最初的命中率包括3 部分:胺、中心氨基酸和N 端。这些CCR1 数据揭示了:①胺部分的改变是不合适的;②分子的中心部位更偏向于容纳小的氨基酸;③N 端的取代基大体变形性较好。这导致CCR1 比其他的CCR 族有更高的选择性和微粒体稳定性,并且具有更好的药物代谢活性。此研究为未来抗癌及消炎药物的发展奠定了基础。
2.1.3 抗癌作用异常蛋白质磷酸化经常引发形成人类各种疾病。在518 个人类基因编码蛋白激酶中,细胞周期素依赖性激酶(CDKs)和糖原合成激酶(GSKs)这两中激酶由于在重大疾病中频频异常,而引起了广泛的关注。CDK 和GSK3 抑制剂的研究已经被视为是一种有效治疗的手段。近年来,大量抑制CDK 和GSK-3β的不同结构一直有报道。只有少量的分子在当前作为CDK 和GSK3 抑制剂。Rajaa Boulahjar 等[23]采用一系列的方法制备出了四氢吡啶并[1,2-a]异吲哚酮衍生物(valmerins),具体反应路线见图15。并调查了其中一种valmerins 在小鼠体内的功效,结果显示,valmerins 阻止了肿瘤的生长而使实验小鼠表现出较好的承受力。valmerins 将促进抗癌药物在未来的进一步发展。
此外,苯并噻唑2-甲酸还可以用来合成治疗神经心血管疾病或障碍的药物[24];作为细胞内钙调节器的组成单元可以用来合成助消化药物[25];还可以用于肥胖症的治疗[26]。

图15 四氢吡啶并[1,2-a]异吲哚酮衍生物的合成Fig.15 Synthesis of valmerins
2.2 材料科学领域的应用
苯并噻唑2-甲酸不仅具有药理活性,其自身是一种具有刚性平面结构以及拥有离域大π键的分子,因此还可以用来作光学材料[27-28]以及荧光材料[29]。例如:苯并噻唑2-甲酸作为中间体,可以制备光学薄膜,并且该类薄膜可以作为平板显示器的贴膜;运用苯并噻唑2-甲酸的荧光性质,可以制备荧光金属离子指标器。
3 结束语
苯并噻唑2-甲酸作为重要的有机医药中间体,其可以应用于抗癌、杀菌消炎以及抗病毒感染等医学研究领域;同时作为含氮杂环化合物,它具有刚性的平面结构以及离域大π键,这些独特的性质使其在光学材料以及荧光材料等材料科学方面得到广泛的应用。
目前,合成苯并噻唑2-甲酸的方法主要有四大类:二氧化碳羧化法、高锰酸钾氧化法、酯水解法、Phillips 合成法。但是,这些方法大多存在步骤较繁琐、条件较苛刻、环境友好性较差等缺点。因此,对苯并噻唑2-甲酸合成方法的改进,还有待进一步探索和研究。
基于苯并噻唑2-甲酸在医学、医药、材料科学等方面的广泛应用,可以预测其未来将可能具有很大的应用价值。
[1] Chevrie D,Lequeux T,Demoute J P,et al. A convenient one-step synthesis of fluoroethylidene derivatives[J].TetrahedronLett,2003,44(44):8127-8130.
[2] Facchinetti V,Gomes C R B,de Souza M V N,et al.Perspectives on the development of novel potentially active quinolones against tuberculosis and cancer[J]. Mini-Reviews,2012,12(9):866-874.
[3] Rahn N,Dunkel R,Kunz K,et al. Goergens:WO,20-10149308[P].2010-12-29.
[4] 李炎,王玉炉.2-取代苯并噻唑的合成进展[J].有机化学,2006,26(6):878-884.
[5] Oleg Vechorkin,Nathalie Hirt,Hu Xile.Carbon dioxide as the C1 source for direct C—H functionalization of aromatic heterocycles[J].Org Lett,2010,12(15):3567-3569.
[6] Ine I F Boogaerts,Steven P Nolan.Carboxylation of C—H bonds using N-heterocyclic carbene gold(I)complexes[J].AM Chem Soc,2010,132(26):8858-8859.
[7] Ine I F Boogaerts,George C Fortman,Marc R L Furst,et al. Carboxylation of N—H/C—H bonds using N-heterocyclic carbenecopper(I)complexes[J]. Angew Chem,2010,49:8674-8677.
[8] Hiroshi Inomata,Ken ichi Ogata,Shin-ichi Fukuzawa,et al.Direct C—H carboxylation with carbon dioxide using 1,2,3-triazol-5-ylidene copper(I)complexes[J]. Org Lett,2012,14(15):3986-3989.
[9] Kamps Jos J A G,Belle Roman,Mecinovic Jasmin. Hydroxylamine as an oxygen nucleophile:Substitution of sulfonamide by a hydroxyl group in benzothiazole-2-sulfonamides[J]. Organic & Biomolecular Chemistry,2013,11(7):1103-1108.
[10]Vamsi Krishna K,Sandala Anuradha Bai,Garige Anil Kumar,et al. Synthesis of new benzothiazolopiperazines/piperidines as antiinflammatoryagents[J].Indian Journal of Heterocyclic Chemistry,2013,22(3):229-232.
[11] Walker Daniel P,Wishka Donn G,Corbett Jeffrey W,et al. Preparation and formulation of N-quinuclidinyl-heteroaryls as nicotinic acetylcholinergic receptor modulators for the treatment of a variety of central nervous system disorders:WO,2002100858[P].2002-12-19.
[12]Qiao Jennifer X,Hu Carol Hui,Wang Tammy C.Quinolinone-carboxamides as inhibitors of endothelial lipase and their preparation:US,2013048942[P].2013-04-04.
[13]罗波,高锦明,王惠.苯并恶唑和苯并噻唑类化合物的合成及其抗菌活性研究[D]. 杨凌:西北农林科技大学,2012.
[14]杨晓亮,苏为科.2-取代苯并噻唑的绿色合成方法研究[D].温州:温州大学,2010.
[15] Glossop Paul Alan,Palmer Michael John,Andrews Mark David.Preparation of aryl substituted carboxamide derivatives as TRPM8 modulators:UK,2012120398[P].2012-09-13.
[16]Ting Pauline C,Aslanian Robert,Cao Jianhua,et al.Preparation of substituted amide derivatives as DGAT-1 inhibitors:US,2012024179[P].2012-02-23.
[17]Morita Mikio,Watanabe Shuzo.Preparation of acylpiperazinylpyrimidine derivatives for use as TTX-S channels blockers:JP,2012020567[P].2012-02-16.
[18] Allen Jennifer R,Chen Jian J,Frohn Michael J,et al.Preparation of nitrogen heterocyclic compounds useful as 3’,5’-cyclic nucleotide-specific phosphodiesterase 10(PDE10)inhibitors:US,2011143365[P].2011-11-17.
[19] Pillaiyar Thanigaimalai,Sho Konno,Takehito Yamamoto,et al.Design,synthesis,and biological evaluation of novel dipeptide-type SARSPCoV 3CL protease inhibitors:Structure-activity relationshipstudy[J]. European Journal of Medicinal Chemistry,2013,65:436-447.
[20] Pillaiyar Thanigaimalai,Sho Konno,Takehito Yamamoto,et al. Development of potent dipeptide-type SARS-CoV 3CL proteaseinhibitors with novel P3 scaffolds:Design,synthesis,biologicalevaluation,and docking studies[J].European Journal of Medicinal Chemistry,2013,68:372-384.
[21]Terasaka Tadashi,Matsuda Hiroshi,Ito Shinji,et al.Preparation of (phenoxy)phenylalkanoic acid derivatives as CRTH2 antagonists for treatment of inflammatory diseases:JP,2011088826[P].2011-05-06.
[22]Cullen L Cavallaro,Stephanie Briceno.Discovery and lead optimization of a novel series of CC chemokine receptor 1(CCR1)-selective piperidine antagonists via parallel synthesis[J].J Med Chem,2012,55:9643-9653.
[23]Rajaa Boulahjar,Aziz Ouach,Chiurato Matteo,et al.Novel tetrahydropyrido[1,2-a]isoindolone derivatives (Valmerins):Potent cyclin-dependent kinase/glycogen synthase kinase 3 inhibitors with antiproliferative activities and antitumor effects in human tumor xenografts[J]. J Med Chem,2012,55:9589-9606.
[24]Blackburn Christopher,Gigstad Kenneth M,Harrison Sean J,et al. Preparation of substituted hydroxamic acids as HDAC6 inhibitors:US,2011106632[P].2011-09-01.
[25] Whitten Jeffrey P,Pei Yazhong,Cao Jianguo,et al. Thiophene derivatives as modulators of intracellular calcium:US,20110065724[P].2011-03-17.
[26]Kawano Tomoaki,Yonetoku Yasuhiro,Hanazawa Takeshi,et al.Preparation of diacylethylenediamine compounds as DGAT1 inhibitors:JP,2010122968[P].2010-10-28.
[27] Okawa Haruki,Piao Mia Bravo,Ochiai Koshiro,et al.Manufacture of compounds for imparting the polarizing property changeable over a wide range of wave lengths to optical films and optical films prepared by using the compounds:JP,2011207765[P].2011-10-20.
[28] Okawa Haruki,Park Meia Brab,Kobayashi Tadahiro,et al.A compound,an optical film containing the same used in flat panel display and method for manufacturing the optical film:JP,101838264[P].2010-09-24.
[29] Gee Kyle,Martin Vladimir. Preparation of benzoxazolebased fluorescent metal ion indicators:US,2010078333[P].2010-07-08.
猜你喜欢
食品科学技术学报(2022年6期)2022-12-15
化工与医药工程(2022年3期)2022-08-08
高中数理化(2022年2期)2022-02-22
世界农药(2019年4期)2019-12-30
世界农药(2019年3期)2019-09-10
天然产物研究与开发(2018年10期)2018-11-06
中学生数理化·高二版(2016年3期)2016-12-26
天然产物研究与开发(2016年11期)2016-06-15
郑州大学学报(工学版)(2015年1期)2015-03-24
影像科学与光化学(2014年3期)2014-03-11
