
硝基脲类含能材料的合成及性能研究进展
2015-05-10 00:44徐志斌孟子晖崔可建刘文进
含能材料 2015年4期
王 瑞, 徐志斌, 孟子晖, 崔可建, 刘文进
(北京理工大学化工与环境学院, 北京 100081)
1 引 言
现代武器装备对含能材料尤其是炸药的热稳定性、撞击感度及摩擦感度等性能有越来越高的要求,而硝基脲类含能材料通常具有以下优点[1-3]: 第一,硝基脲类化合物因存在内在高密度的脲结构而具有较高密度。一般而言,硝基脲类含能材料的密度大于1.90 g·cm-3; 第二,双硝基脲类化合物属于富氧型分子,极大地促进了该类化合物的氧平衡; 第三,单硝基脲类化合物有利于分子间氢键的生成,是钝感含能材料的良好选择; 第四,双硝基脲类化合物易水解,可得到开环的双硝胺化合物,作为合成其它含能材料的前体。相对而言,单硝基脲类化合物不易水解且较钝感。然而,双硝基脲类含能材料水解稳定性较差,限制了其应用。因此为了弥补硝基脲类含能材料的不足,设计合成高能、钝感、稳定性好的硝基脲类含能材料迫在眉睫。2013年孟子晖教授课题组报道了四硝基甘脲二聚体(TNDGU)的性能[4-5],TNDGU爆轰性能与RDX相当,感度低于RDX,密度高于RDX,稳定性和安全性好,具有较好的综合性能。
基于以上优点,对新型硝基脲类含能材料的合成探索研究已成为含能材料领域研究的热点。因具有不同元环的硝基环脲类化合物在结构稳定性和环张力上存在差异,因而相应化合物的合成方法及性能也有所区别。本文将硝基脲类含能材料分为硝基环脲类含能材料和非环硝基脲类含能材料两大类,并重点介绍硝基环脲类含能材料的合成及性能研究进展,为下一步高能钝感硝基脲类含能化合物的设计与合成提供借鉴。
2 硝基五元环脲类含能材料的合成
2.1 以脲为原料
2.1.1 以甘脲衍生物为中间体
目前有关甘脲衍生物应用于含能材料领域的合成研究相对较少,甘脲所具有的特殊结构增加了其密度和稳定性,有望成为良好的含能材料前体。
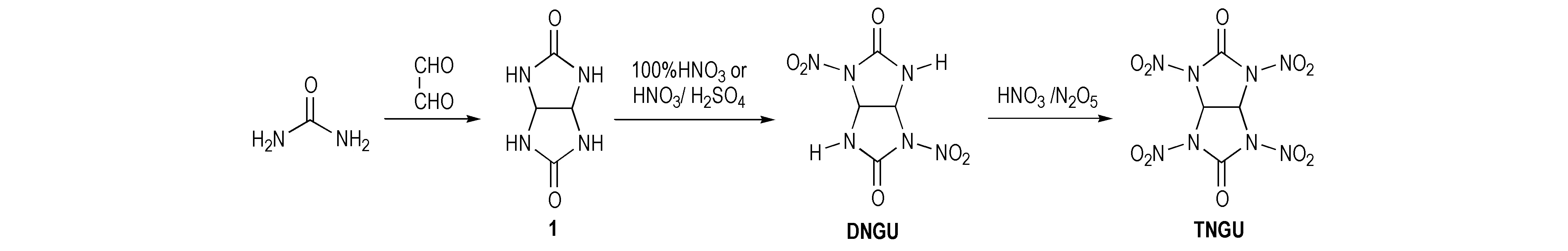
1965~1966年万道正等[6]首先使用甘脲(1)为原料在0~5 ℃下一步硝化法合成出双硝基脲炸药1,3,4,6-四硝基甘脲(TNGU),当时合成过程未公开。1974年,法国化学家Boileau[7-8]首次公开报道了硝脲炸药1,4-二硝基甘脲(DNGU)和1,3,4,6-四硝基甘脲(TNGU)的合成。即以脲和乙二醛为原料环缩合生成甘脲(1),再用100%HNO3或HNO3/H2SO4硝化制得DNGU,最后用HNO3/N2O5作硝化剂合成出TNGU,如Scheme 1所示。该法原材料便宜易得,反应步骤简单,但TNGU即使在冷水中也很容易水解[9],需要用到硝化能力较强的无水硝化剂HNO3/N2O5。易文斌等[10-11]对该合成路线进行优化,用100%HNO3/Ac2O作硝化剂硝化DNGU生成TNGU,得率较高,为63%,该法合成条件温和,操作简便,重现性高。
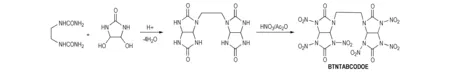
双硝基脲化合物TNGU易水解的特性限制了其应用,为了合成高密度、高能量和稳定性好的化合物,可考虑将双硝基脲转换成单硝基脲,同时不减少致爆基团的数目。甘脲多聚体的硝基衍生物——四硝基甘脲二聚体(TNDGU)[4-5]的合成解决了这一问题,既降低了感度,又提高了水解稳定性。TNDGU以甘脲为原料,通过多聚甲醛二聚得到甘脲二聚体(GD),然后用HNO3/Ac2O硝化所得,合成路线见Scheme 2。方银高等[12]通过甘脲羟甲基化再与硝仿反应引入两个高能量的硝仿基团,再硝化得到 1,3-二硝基-2,4-双(三硝基乙基)-甘脲(BTNDNG),胡荣祖等[13]报道了1,5-二甲基-2,6-双(三硝乙基)-4,8-二硝基甘脲(DMBTNTDNGU)的合成及部分性能,其合成路线见Scheme 3。

Scheme 1[8]

Scheme 2[5]

Scheme 3[12-13]
Scheme 4[14]
对甘脲类含能化合物,可从两方面着手研究: 一是对其中合成工艺不佳的已知化合物进行工艺优化,通过提高其反应产率以达到降低生产成本,实现工业化生产和应用的目的; 二是以甘脲或甘脲多聚体等为结构单元设计良好的氮杂环骨架分子,引入一些含能取代基如硝基、硝仿基、叠氮基和氨基等进行修饰,进一步提高甘脲类含能材料的能量水平和稳定性。
2.1.2 以咪唑烷-2-酮类衍生物为中间体
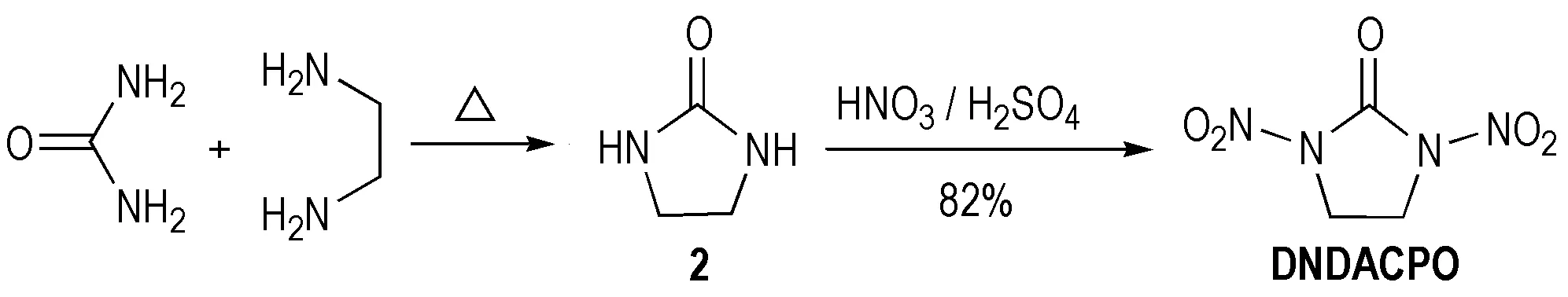
1,3-二硝基-1,3-二氮杂环戊酮(DNDACPO)以脲和乙二胺为原料,经环缩合生成咪唑烷-2-酮(2)中间体,然后经硝硫混酸硝化所得[15-16],产率为82%,合成路线见Scheme 5。后来Kuchurov等[17]用N2O5于液态CO2中硝化咪唑烷-2-酮(2)得到DNDACPO,产率为93%。与传统的硝化过程相比,该方法硝化较为容易,产率高,而且潜在爆炸的危险较低,反应过程基本无污染,属于环境友好型硝化剂,可以说在硝化剂方面有一个重大突破性进展。
Scheme 5[15-16]
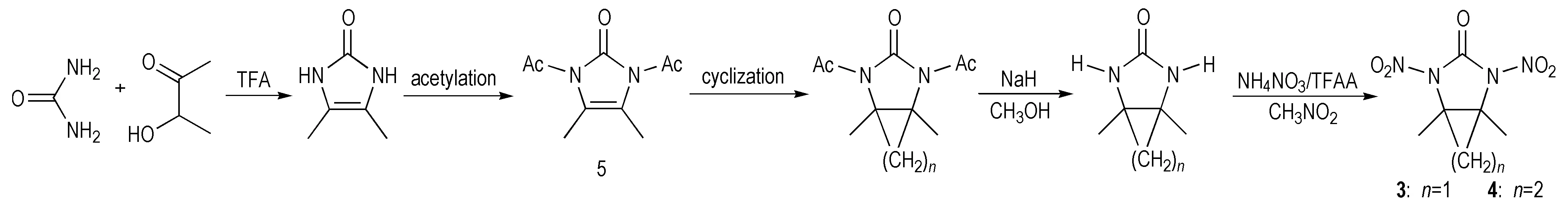
五元单环硝基脲化合物爆速和能量均较低,Gagnon等[18]通过烷基化反应合成出了双环化合物1,5-二甲基-2,4-二硝基-2,4-二氮杂双环[3.1.0]已烷-3-酮(3)和1,5-二甲基-2,4-二硝基-2,4-二氮杂双环[3.1.0]庚烷-3-酮(4) ,合成路线如Scheme 6所示。化合物3和化合物4均以1,3-二乙酰基-4,5-二甲基-4-咪唑啉-2-酮(5)为前体。其环丙烷化反应需在无水环境中进行,可加乙酰氯除掉其中的痕量水,同时体系中产生的氯化氢也起到了加速反应和提高产率的作用,随后NaH脱酰基再经NH4NO3/(CF3CO)2O硝化得到化合物3。化合物4的合成路线与化合物3不同之处在于,其环丁烷化是用乙烯光催化所得。
Scheme 6[18]
化合物3和化合物4因存在环张力较大的三元环和四元环导致其稳定性较差,较难应用于含能材料领域。Pagoria等[1]用含氮五元杂环与脲反应合成出了2,4,6,8-四硝基-2,4,6,8-四氮杂双环[3.3.0]辛烷-3-酮(K-55)和2,4,6-三硝基-2,4,6, 8-四氮杂双环[3.3.0]辛烷-3-酮(HK-55),在对2,4,6,8-四氮杂双环[3.3.0]辛烷-3-酮二盐酸盐(6)进行硝化时,发现用90%HNO3/Ac2O在低于10 ℃硝化时,得到HK-55,产率72%; 而用100%HNO3/Ac2O在20~50℃时硝化得到K-55,产率49%,反应路线见Scheme 7。
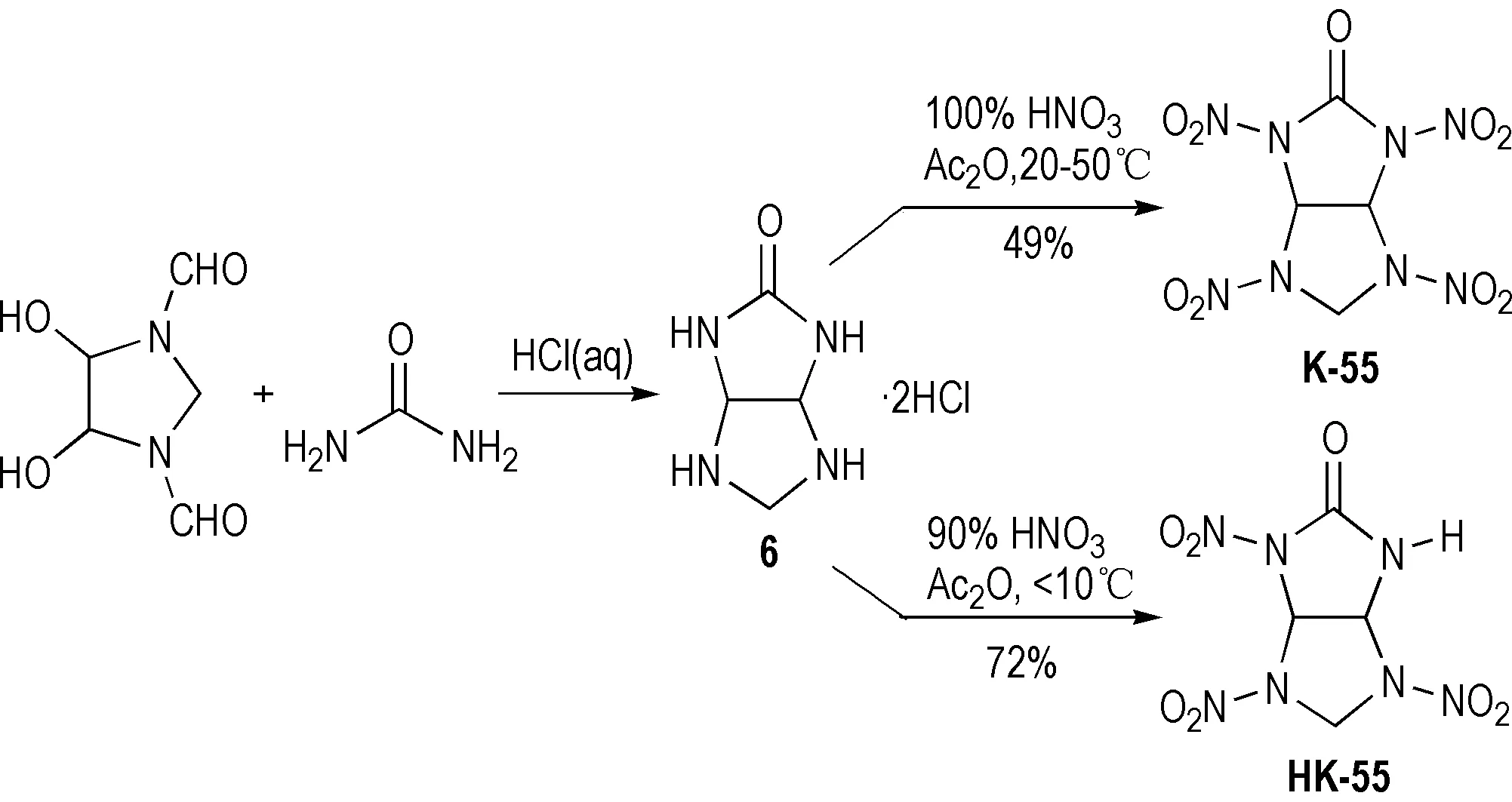
Scheme 7[1]
为进一步提高硝基环脲类化合物的稳定性,人们以稳定性较高的六元氮杂环和脲为原料合成出了2,5,7,9-四硝基-2,5,7,9-四氮杂双环[4.3.0]壬烷-8-酮(K-56)。K-56与TNGU相比少了一个羰基,但亚甲基比羰基有较好的供电性与稳定性,这会使K-56性能发生一定的变化。对于K-56的合成,Graindorge等[19]以乙二胺与甲酸甲酯为原料,经N-甲酰化、醛胺缩合,与脲反应闭环得咪唑烷-2-酮衍生物(7),最后经HNO3/ N2O5硝化得K-56,合成路线见Scheme 8。后来Agrawal等[20]简化了合成K-56过程,以乙二胺和乙二醛为原料,经缩合反应再与脲一锅煮得到硝化前体,随后用HNO3/Ac2O硝化得到K-56,产率82%,合成路线见Scheme 9。
Boyer及其团队[21]报道了2,4,6,8,10,12-六硝基-2,4,6,8,10,12-六氮杂三环[7.3.0.03.7]十二烷-5,11-二酮(HHTDD)的合成,其母体盐酸盐(8)是用1,4-二甲酰基-2,3,5,6-四羟基哌嗪(DFTHP)与两分子脲在酸性条件下反应所得,然后经HNO3/P2O5硝化得到HHTDD,产率为74%,如Scheme 10所示。同时Boyer对硝化反应作了详细研究,发现纯HNO3低温硝化仅能硝化哌嗪上的氮得到化合物9,产率为28%; 脲结构基团上的氮硝化较为困难,用HNO3/Ac2O硝化可得到不完全硝化产物10和11,若用额外的HNO3/Ac2O或HNO3/(CF3CO)2O处理则可得到五硝基衍生物12; 用NO2BF4/CH3CN处理化合物10,11和12可得到HHTDD。HHTDD在酸性条件下不稳定,N—C键断裂分解生成2-氧代-1,3-二硝基咪唑并[4,5-b]吡嗪(13)[22],产率为17%,反应式见Scheme 11。这对其它化合物设计与合成有一定的参考价值。
2.2 以胍类衍生物为原料
此合成方法与前法的不同之处在于: 硝化前体均为胍类衍生物,即以胍或硝基胍与氮杂环化合物反应环化所得,然后经硝化得到硝基环脲类化合物。此方法是间接合成硝基脲类化合物的一种重要方法,在强氧化性硝化剂中,胍基很容易被氧化生成羰基。
Mckay等[23]以胍类衍生物为原料合成出了1,3-二硝基-1,3-二氮杂环戊酮(DNDACPO),即以2-硝氨基-1,3-二氮杂环戊烯(14)为原料,经HNO3硝化得到单硝基取代的硝基胍衍生物(15),重排后结构变为1-硝基-2-硝亚氨基咪唑烷(16),继续加入硝酸,硝亚氨基被氧化成羰基而得到DNDACPO,产率为86.4%,反应路线如Scheme 12所示。Boyer等[24]报道了胍基三环化合物17的合成,在浓盐酸中1,4-二甲酰基-2,3,5,6-四羟基哌嗪(DFTHP)与2摩尔当量的胍缩合成环所得。将其用100% HNO3/Ac2O硝化则可得到HHTDD,反应式见Scheme 13。此法与以脲为原料(见Scheme 10)合成相比,反应时间长,且产率相对较低,但对于新型硝基脲类化合物的合成具有一定的研究意义。

Scheme 8[19]
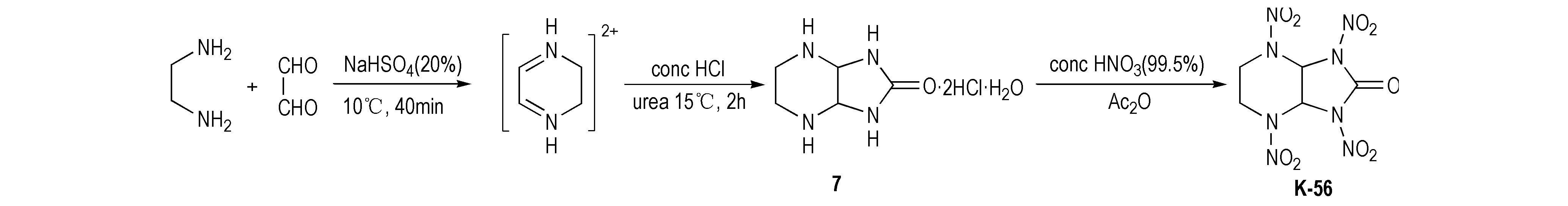
Scheme 9[20]
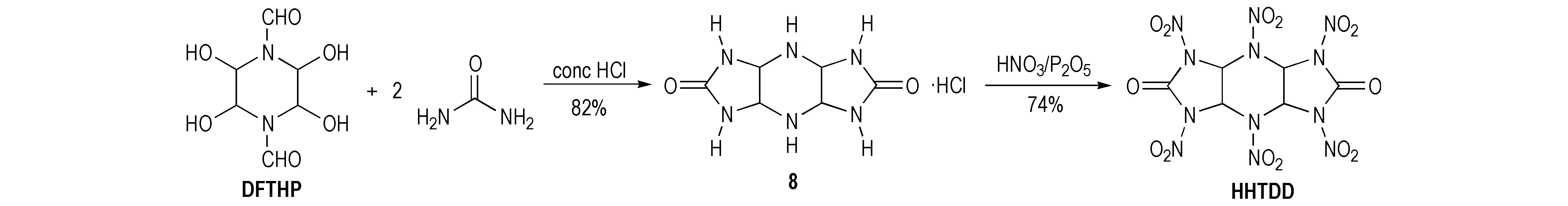
Scheme 10[21]

Scheme 11[22]
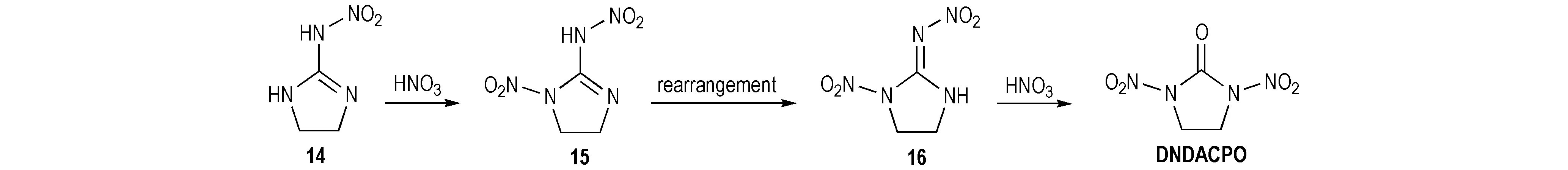
Scheme 12[23]

Scheme 13[24]
2.3 以其它小分子为原料
此合成方法不同于前两种方法,环脲中间体是用非脲非胍小分子反应所得,即以胺或不饱和氮杂环与其它小分子环缩合而成,再脱去保护基或直接硝化制得硝基脲类化合物。由于小分子结构选择的灵活性和合成方法的多样性,对具有特殊结构或爆轰性能较好的化合物合成具有重要的研究意义。
Pagoria等[1]以化合物18为原料经溴代后,再与乙撑二硝胺(EDNA)在三乙胺盐作用下反应生成7,9-二乙酰基-2,5-二硝基-2,5,7,9-四氮杂双环[4.3.0]-8-酮(19)和7-乙酰基-2,5-二硝基-2,5,7,9-四氮杂双环[4.3.0]-8-酮(20),此混合组分说明了N,N′-二乙酰基取代化合物在乙腈的胺溶液里容易脱去保护基而生成了N-单酰基取代化合物,再经20%N2O5/HNO3硝化得到N-乙酰基-N-硝基环脲中间体(21),产率为80%,最后用三氟甲基磺酸酐TFMSAA/20%N2O5/HNO3体系硝化得到化合物K-56,产率为69%,具体合成路线如Scheme 14所示。该法合成路线稍长,而且反应复杂,不适合推广应用。然而值得一提的是,N-乙酰基-N-硝基环脲中间体(21)的乙酰基很难硝解,在20~50℃采用20%N2O5/HNO3、Ac2O/20%N2O5/HNO3和TFAA/100%HNO3,以及在20~40℃采用TFAA/20%N2O5/HNO3等进行硝解时,均未发生反应。
Fischer等[25]合成出了环丁烷的硝胺基衍生物1,3,4,6-四硝基八氢环丁基-[1,2-d:3,4-d′]二咪唑基-2,5-二酮 (TDCD)。首先用2,2-二乙氧基乙胺和氰酸钾酸性条件下生成脲衍生物(22),经环化制得咪唑啉酮中间体(23),再经乙酰化和光催化诱导[2+2]环加成得到环丁烷衍生物(24),弱碱性条件下脱酰基后再硝化得到TDCD,反应路线如Scheme 15所示。
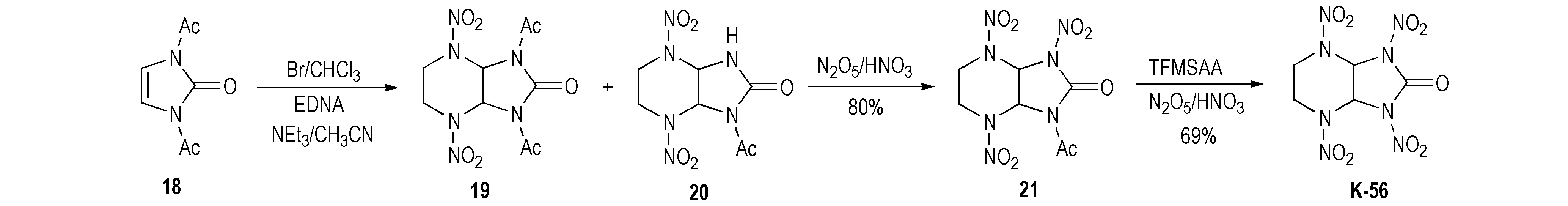
Scheme 14[1]
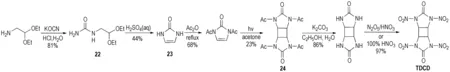
Scheme 15[25]
3 硝基六元环脲类含能材料的合成
与硝基五元环脲类化合物相比,硝基六元环脲类化合物种类较少,爆轰性能较好的化合物主要有2,4,6-三硝基-2,4,6-三氮杂环已酮(K-6)、四硝基丙烷二脲(TNPDU)和2,4,8,10-四硝基-2,4,8,10-四氮杂螺[5.5]十一烷-3,9-二酮(TTUD)等三种,其中K-6的合成研究得很成熟,产率和纯度均较高,并对以硝基胍为原料合成K-6的机理进行了推断。
3.1 以脲为原料
Mitchell等[26]以脲、甲醛和叔丁胺为原料,通过Mannich缩合反应合成出5-叔丁基-1,3,5-三氮杂环己酮(TBT)作为硝化前体,采用HNO3/N2O5/Ac2O、HNO3/N2O5/(CF3CO)2O、NO2BF4/CH3CN等硝解剂来合成2,4,6-三硝基-2,4,6-三氮杂环已酮(K-6),发现在NO2BF4/CH3CN中制备K-6的产率最高,为61%,其合成路线如Scheme 16所示。该法起始原料易得价廉,操作安全方便,但由于前体缩合物TBT的产率仅60%左右,致使反应总产率较低,第二步硝化反应还发生一些副反应,产物不纯。

Scheme 16[26]
周诚等[27]对该合成工艺进行了优化研究,使K-6的总产率提高至48.0%,纯度达98.8%,且不含RDX杂质。但从原子经济性角度看,TBT作为前体合成K-6不经济。
Sikder等[20]以乙烯乙烷基醚和三乙氧基甲烷为原料合成四乙氧基丙烷(25),然后在强酸性条件下与脲缩合成环,再硝化生成四硝基丙烷二脲(TNPDU),其合成路线见Scheme 17。由于比TNGU多了一个亚甲基,TNPDU因具有六元环结构而显得愈加稳定。
3.2 以胍类衍生物为原料
Mckay等[23]研究了部分环硝基胍类化合物的硝化产物。2-硝氨基-1,3-二氮杂环已烯(26)经HNO3硝化得到单硝基取代的1-硝基-2-硝氨基-1,3-二氮杂环已烯(27),该化合物不稳定,经重排后其结构变为1-硝基-2-硝亚氨基-1,3-二氮杂环已烷(28),继续加入硝酸,硝亚氨基被氧化成羰基而得到1,3-二硝基-1,3-二氮杂环已烷(29),反应路线见Scheme 18。1,3-二硝基-4-甲基环已酮(30)以2-硝氨基-4-甲基环已-2-烯(31)为原料用7摩尔当量HNO3/Ac2O硝化所得; 若硝化剂量不足时则生成单硝基化合物32和33[28],反应式见Scheme 19。2-硝氨基-5-羟基-1,3-二氮杂环已-2-烯(34)经HNO3/Ac2O硝化得到1,3-二硝基-5-硝酸酯-1,3-二氮杂环已酮(35),用HNO3或HNO3/H2SO4硝化时仅得到硝酸酯化合物(36),反应式见Scheme 20。
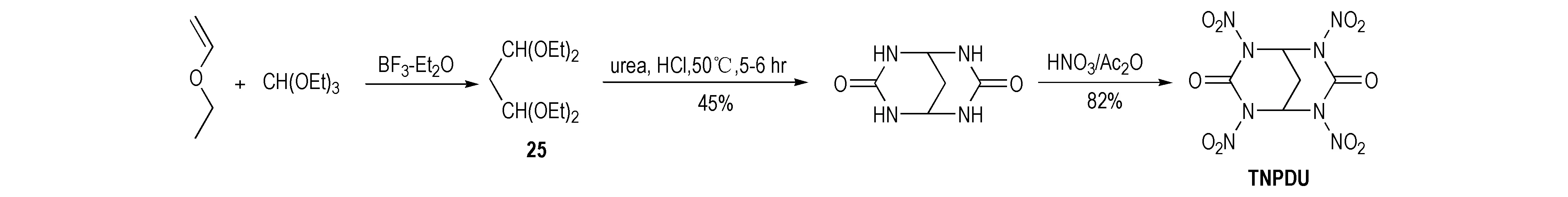
Scheme 17[20]
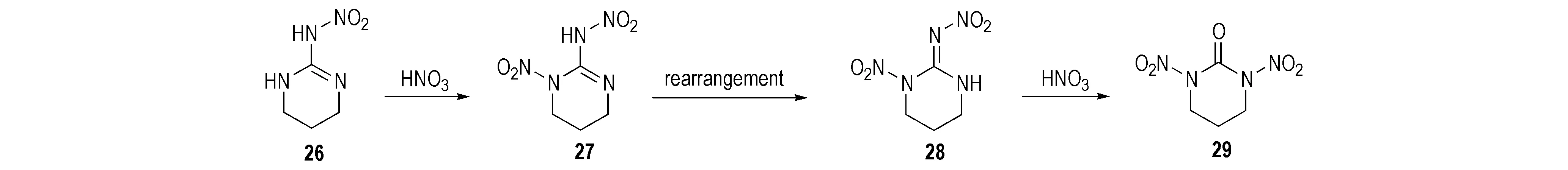
Scheme 18[23]
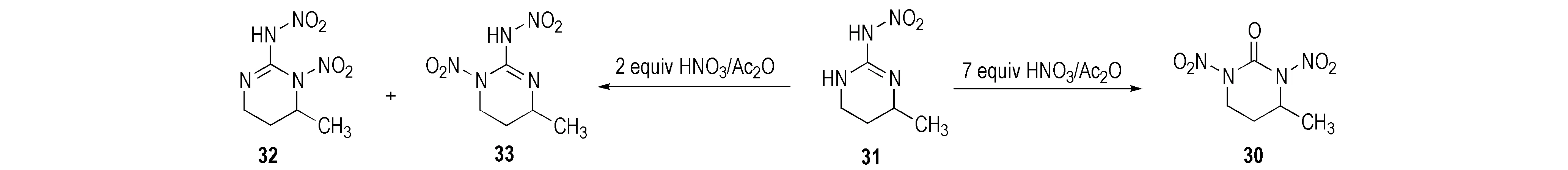
Scheme 19[28]
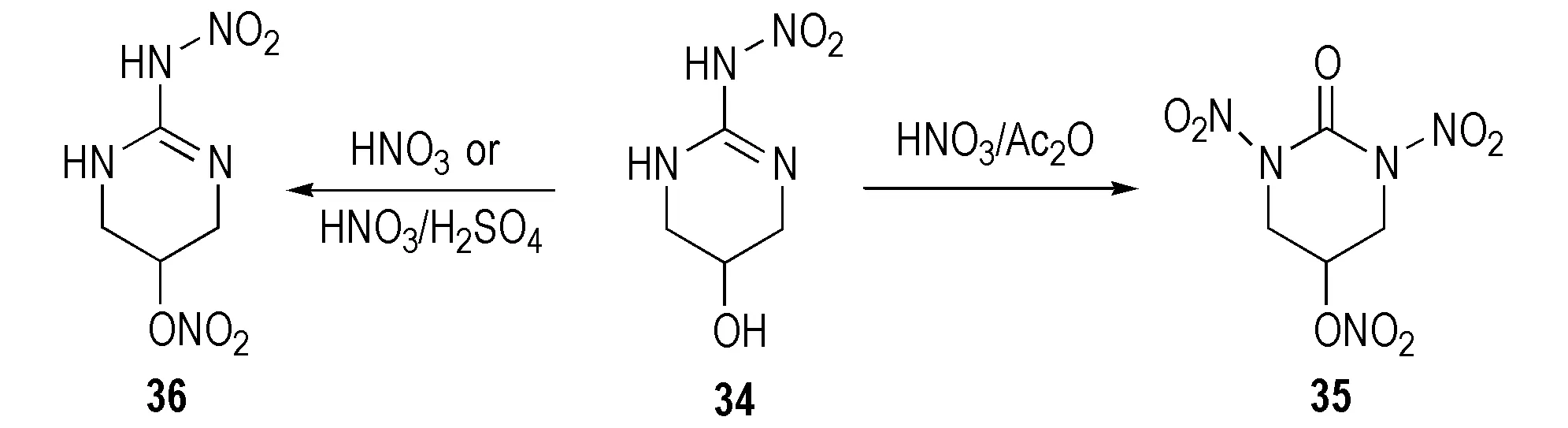
Scheme 20[28]
通过实验结果可知,硝化剂用量直接影响着胍类化合物的硝化产物,硝化剂用量较低时则生成含硝基胍结构的硝化产物,但此类化合物很不稳定,在水中煮沸数分钟后90%以上会被破坏掉,鉴于此,虽然其能量较高,但不适合作为炸药使用; 硝化剂用量较大时硝亚氨基不稳定,易转化成羰基从而生成硝基环脲类化合物。硝化产物与硝化剂种类直接相关,硝化能力强的硝化剂可将其硝化生成硝基脲类化合物。因此,用胍类化合物作反应物时,要注意硝化剂的种类和用量。
Huang等[29-30]使用乌洛托品和硝基胍为原料缩合生成2-硝亚氨基-1,3,5-三氮杂环已烷(37),再与硝酸钠酸性条件制备2-硝亚氨基-5-硝基-1,3,5-三氮杂环已烷(38),然后用HNO3/(CF3CO)2O硝化得到K-6,产率为62.2%,合成路线见Scheme 21。但该法中每步反应持续时间均较长,乌洛托品利用率低; 最后一步硝化需用(CF3CO)2O,这明显增加了K-6的制备成本。周诚等[31]对此合成工艺进行了优化改进,以乌洛托品和硝基胍为原料,通过Mannich反应得到2-硝亚氨基-1,3,5-三氮杂环已烷盐酸盐(37),经HNO3/Ac2O硝化得到K-6,产率为72.0%,纯度为98.5%,合成路线如Scheme 22所示。与以脲为原料合成相比,这是一种产率高、纯度高、节约时间的新型合成方法。周诚等[31]对此反应进行详细的机理分析后认为,2-硝亚氨基-1,3,5-三硝基-六氢化-1,3,5-三嗪(NTNHT)中硝亚氨基强吸电子基团的引入,造成与之相连的C原子呈缺电子状态,显电正性。水分子进攻Cδ+,生成过渡态T-1,T-1极不稳定,易脱去硝酰胺形成羰基化合物K-6,硝酰胺亦不稳定,分解生成N2O与H2O,反应机理如Scheme 23所示。
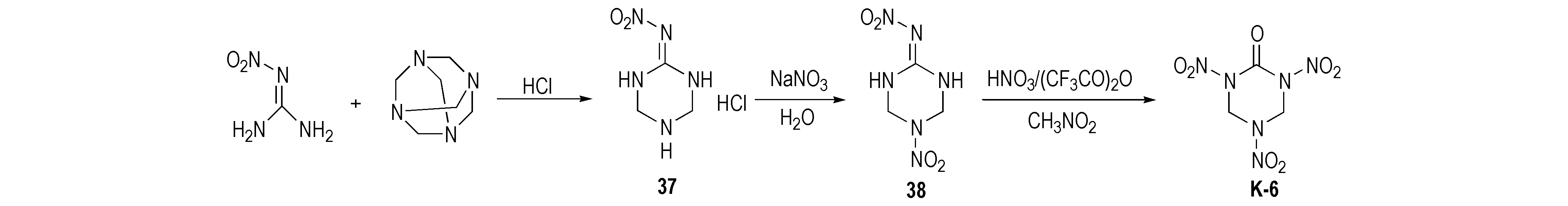
Scheme 21[30]

Scheme 22[31]
符全军等[32]以季戊四胺二硫酸盐为原料,与硝基胍环缩合生成3,9-二硝亚氨基-2,4,8,10-四氮杂螺[5.5]十一烷(39),再经HNO3/Ac2O硝化得到螺环硝基脲类化合物2,4,8,10-四硝基-2,4,8,10-四氮杂螺[5.5]十一烷-3,9-二酮(TTUD),总产率为45%,合成路线见Scheme 24。若将硝化剂改为HNO3/H2SO4或HNO3/P2O5体系硝化,硝化产率均在90%以上。
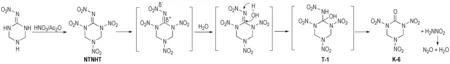
Scheme 23[31]

Scheme 25[32]
3.3 以其它小分子为原料
符全军等[32]以季戊四胺二硫酸盐为原料,考虑与其它小分子环缩合生成螺环化合物硝化前体,实验发现,与CS2环缩合生成四氮杂螺环硫化物(40),再用HNO3/Ac2O硝化可得到螺环化合物TTUD,但产率较低,仅为6%,这是因为在反应过程中体系有单质硫和连硫产物等高分子聚合物产生,包裹在反应物表面,阻止反应顺利进行。有机化合物分子中的硫脲基在氧化剂存在下易转变成脲基,虽然该合成路线产率较低,经济性不高,但对于新型硝基脲类化合物的合成研究具有一定的参考价值。
4 硝基七元环脲类含能材料的合成
文献调研结果表明,目前已有合成报道的硝基七元环脲化合物仅有一个,即1,3-二硝基-1,3-二氮杂环庚烷-2-酮(41),且只有其合成报道,无爆炸性能数据。化合物41以2-硝氨基-1,3-二氮杂环庚-2-烯(42)为原料经99%HNO3/Ac2O硝化所得[23],产率85.4%,反应式见Scheme 26。当硝化剂用量较大时,2-硝氨基-1,3-二氮杂环庚烯很容易硝化生成硝基环脲化合物,低温硝化则生成硝胺基硝化产物,该物质很不稳定,在水中煮沸5 min 97%就会被破坏掉,不适合作为炸药使用。

Scheme 26[23]
综上,所有硝基环脲类化合物中,五元环脲类化合物研究得较充分,尤以咪唑烷-2-酮为原料合成研究较多。甘脲因内部含有高密度的脲基团结构而成为合成高能量密度化合物良好的骨架,可将甘脲或甘脲多聚体为结构单元进行基团修饰或衍生; 六元环脲化合物报道较少,因其具有结构稳定性好的六元环可作为结构单元去构建新型化合物; 七元环脲和八元环脲类化合物亟待开发,当以脲为原料反应得不到目标产物时,可考虑用含亚硝基或硝基胍类或其它小分子等化合物进行合成探索。化合物中脲基团上的氮,相对于其它环(如吡嗪环等)上的氮硝化要困难一些,因此可根据实际需要选择不同硝化能力的硝化剂。
5 非环硝基脲类含能材料
非环硝基脲类含能材料是指分子中脲基团氮上氢被硝基取代而生成的非环化合物。目前这种含能材料较少,已合成出的主要有单硝基脲(NU)和N,N′-二硝基脲(DNU)。目前报道硝基脲(NU)的合成方法有两种[33]。脲和硝酸反应生成硝酸脲(UN)[34],然后用硫酸或乙酸酐/乙酸水解UN,产率分别为13%和67.2%[33],见合成路线Scheme 27。很显然,用乙酸酐/乙酸处理的产物产率高,这是一种较为经济的合成方法。 DNU的合成[35]是以尿素为原料,硝化剂为20%发烟硫酸与100%硝酸(V/V=0.6/1),两阶段的反应温度分别为-15~-10℃和0~5 ℃时硝化合成出DNU,收率可达83.2%,见Scheme 28,比Patrick等[36]在100%硝酸/95%浓硫酸硝化得DNU的39%产率高很多,同时实验证实了DNU是尿素分两步硝化所得,即在硝化过程中先生成了硝基脲(NU),然后进一步生成二硝基脲(DNU)。
DNU因具有高密度(1.98 g·cm-3)、高爆压(36.1 GPa)和高爆速(8861 m·s-1)而表现出较好的爆轰性能[37],但其本身对摩擦和冲击非常敏感,热稳定性和化学稳定性较差,从而限制了其在含能材料领域的直接应用。可将DNU转化为相应的有机盐等高能量离子盐,来克服它的热不稳定性,降低其摩擦感度和冲击感度; 也可作合成双环和笼状杂环N,N′-二硝脲类化合物的重要前体原料[37]。
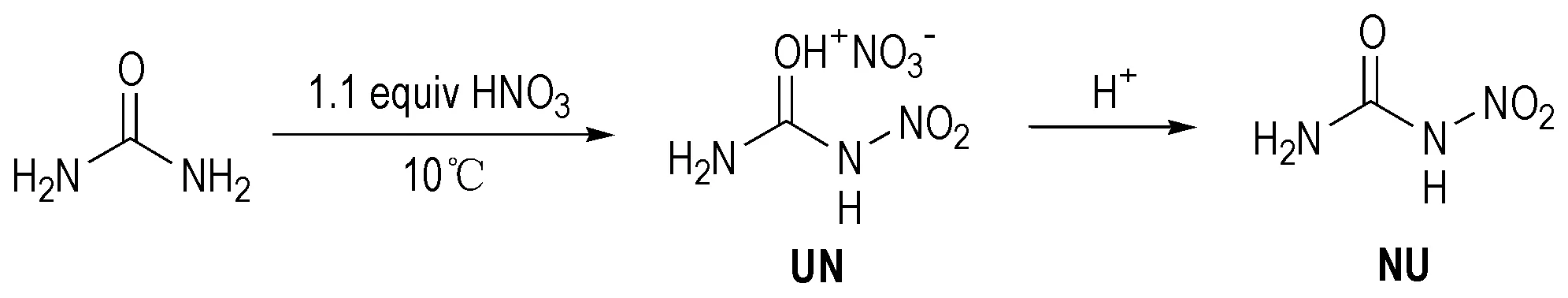
Scheme 27[34]
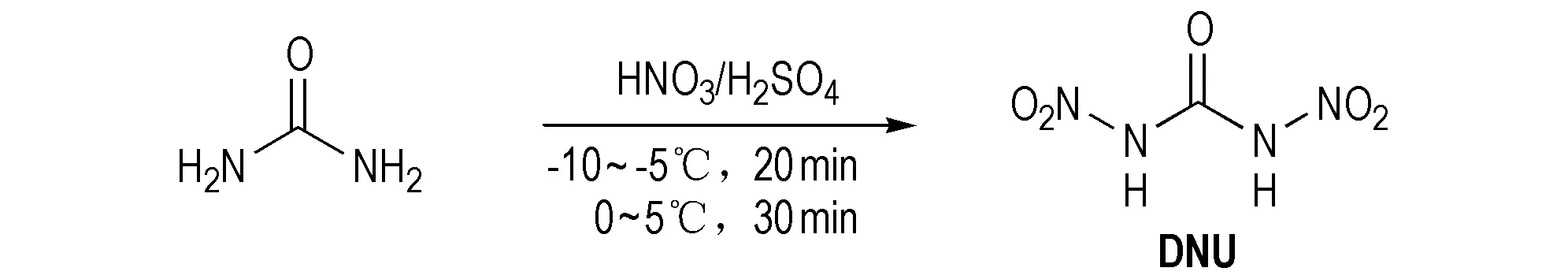
Scheme 28[35]
6 硝基脲类化合物的性能
一种含能材料在进行实际应用和研究其配方前,了解其性能对进一步应用有着至关重要的意义,只有充分了解它的热稳定性、水解稳定性、理化性能及爆轰性能等各项性能,才能在实际应用中发挥最大的作用,为其今后在武器系统中应用提供基础实验依据。
6.1 硝基环脲类化合物的热稳定性
席于烨等[38]通过DSC分析研究了部分硝基环脲类化合物的热稳定性,其中最为人熟知的硝基脲类化合物DNGU和TNGU的热解峰温分别为257 ℃和180 ℃,并发现下面6种硝基脲类化合物(见Scheme 29)的热稳定性顺序为DNGU>TNPDU>DNDACPO>HHTDD>TNGU>化合物43,并实验证实环境气氛对这些物质的热解影响不大。DNGU最稳定说明—H取代—NO2后增加了分子稳定性,TNPDU稳定性次之,说明六元环替代五元环之后较小的环张力增加了分子热稳定性,K-6可以说明这个问题,其因具有六元环结构而表现出较好的热稳定性(DSC放热峰温为211.4 ℃[31])。化合物43热稳定性最差(170 ℃开始分解,192 ℃爆炸性分解)主要归因于四元环张力较大,从而导致该化合物稳定性差,并通过热解引发机理研究发现N—NO2键相对较弱,易成为爆炸或热解的引发键,但是对于环张力较大的硝基脲类化合物,环破裂与N—NO2键存在相互竞争,环优先破裂也是有可能的。
甘脲多聚体的硝基衍生物TNDGU[4]通过DSC和TG-DTG分析可知其热稳定性比较理想,其热分解峰温为284.79 ℃,比DNGU和TNGU具有更加良好的热稳定性,即甘脲二聚结构的形成,对提高化合物结构的热稳定性有很大作用。谭国洪[14]对新型环脲化合物1,2-二[4,6,8-三硝基-2,4,6,8-四氮杂双环[3.3.0]辛二酮-3,7]乙烷(BTNTABCODOE)研究发现,其热解峰值温度为229 ℃,比TNGU(180 ℃)具有更加良好的热稳定性。
对1,3-二硝基-2,4-双(三硝基乙基)-甘脲(BTNDNG)和1,5-二甲基-2,6-双(三硝乙基)-4,8-二硝基甘脲(DMBTNTDNGU)的热稳定性研究[13,39]发现,二者热解峰温值均在210 ℃左右,具有较高的热稳定性,并通过实验证实N—CH2C(NO2)2基团在加热升温过程中优先离去,即N—CH2C(NO2)2的热稳定性比N—NO2差得多,但该化合物爆轰性能良好,也是一种性能良好的高能材料,其分解机理推断见Scheme 30。
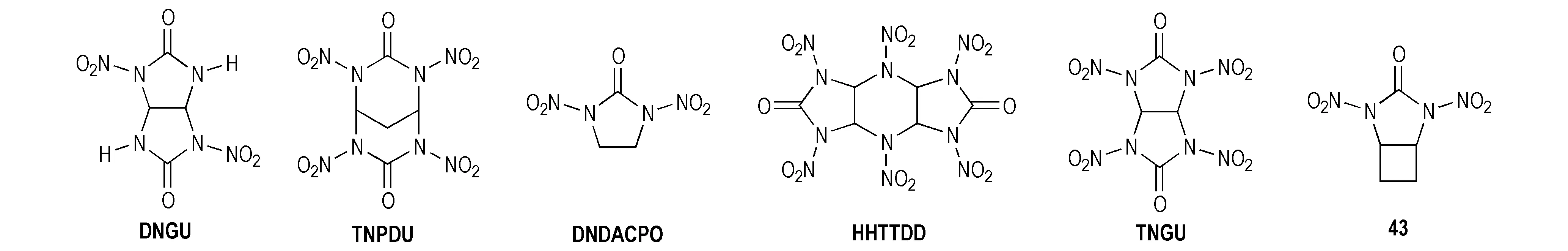
Scheme 29[38]
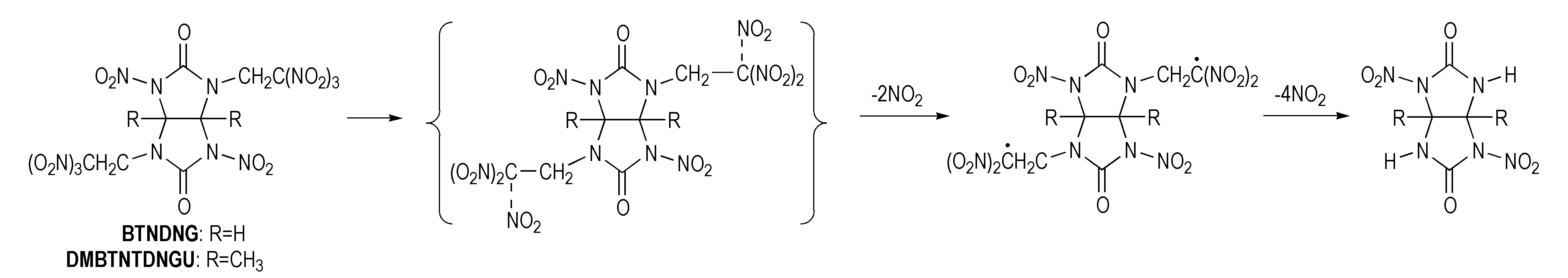
Scheme 30[13,39]
由此可知,一般情况下,单硝基环脲类化合物比双硝基环脲类化合物具有较好的热稳定性,且具有六元环结构的硝基脲类化合物通常热稳定性较好。四硝基甘脲TNGU的热稳定性较差,可对其进行含能取代基修饰或衍生,如甘脲多聚体的硝基衍生物、硝仿基甘脲和烷撑双环脲等物质的稳定性与TNGU相比均有所提高。
6.2 硝基环脲类化合物的水解稳定性
最为人们熟知的单或双硝基环脲类化合物为1,4-二硝基甘脲(DNGU)和1,3,4,6-四硝基甘脲(TNGU),TNGU在冷水中就很容易水解[9],而DNGU用沸水处理仅缓慢分解[40]。二者的水解稳定性从一定程度上说明了单或双硝基环脲类化合物的特性: 双硝基环脲类化合物水解稳定性较差,而单硝基环脲类化合物却有着较好的水解稳定性。
TNGU水解稳定性差的特性限制了其作为含能材料使用,可以考虑将双硝基脲类化合物变成单硝基脲类化合物,同时不减少致爆基团的数目。甘脲多聚体具有特殊的氮杂环结构,其硝基衍生物四硝基甘脲二聚体(TNGDU)[4]属于单硝基脲类化合物,有着较好的水解稳定性。方银高等[12]用三硝基乙基取代TNGU分子中的两个硝基首次合成出1,3-二硝基-2,4-双(三硝基乙基)-甘脲(BTNDNG),与TNGU相比,BTNDNG的水解稳定性有了明显的改善。甘脲衍生物1,2-二[4,6,8-三硝基-2,4,6,8-四氮杂双环[3.3.0]辛二酮-3,7]乙烷(BTNTABCODOE)的水解稳定性与TNGU相比,相同条件下失重量只有后者的1.15%~3.47‰,确实表现出良好的水解稳定性,化合物BTNTABCODOE是非对称取代的新型硝基环脲炸药,改善了其水解稳定性,可作为火炸药配方的组分。
胡荣祖等[42]对多种硝基脲类化合物的水解稳定性进行了详细研究,发现HHTDD对酸和水极其敏感,其环张力的存在也使其水解稳定性较差,并通过实验证明在室温下该化合物宜在相对湿度低于20%的情况下保存,同时对其在中性条件下的水解机理作了初步推断,如Scheme 31所示。研究发现TNPDU的水解稳定性比TNGU要好很多,初步分析原因可能是甲基化,即TNPDU比TNGU多了一个亚甲基而形成稳定性较好的六元环结构,使之具有更好的供电性与稳定性[40]。化合物TTUD的水解稳定性较好,在相对湿度75%和40 ℃下24 h后热失重仅为0.001%,而且TTUD是一种螺环炸药,六元螺环的优势既可引入致爆基团,作为密堆积的骨架分子又可提高其结晶密度,是研究合成新型硝基脲类含能材料的一个新方向[32,42]。

Scheme 31[42]
由此可见,环脲化合物的水解稳定性与紧邻羰基的氮原子上所连基团息息相关。双硝基脲类化合物的水解稳定性均较差,主要是因为硝基在脲基团两旁对称取代,硝基的诱导效应与脲羰基p-π共轭双重作用,引起羰基碳原子电子云转移而显正电性,易受水的亲核进攻而水解,从而致使这类炸药的水解稳定性均较差,难以满足使用要求。单硝基脲类化合物由于脲羰基旁减少一个吸电性的硝基,其碳原子电子云密度会明显增加,从而不易遭受极性水分子的亲核攻击而水解,而且分子内或分子间的氢键使整个分子趋于稳定[13],然而单硝基脲类化合物因为硝基的缺少而使其感度和爆轰性能有所降低。
6.3 环硝基脲类化合物的爆轰性能
表1 部分硝基环脲类化合物的爆轰性能1)
Table 1 Detonation performance of some nitro-cyclourea compounds

entrynamestructuredensity/g·cm-3detonationvelocity/m·s-1impactsensitityH50/cmfrictionsensitity/kgref.1DNDACPO1.79-1.817655——[12]2TNGU2.0492051.862)68.62)[4,41]3DNGU1.8778558872%3)[43-44]4TNDGU1.93830517.0496%3)[4]5BTNDNG1.95903723.5100%3)[12]6DMBTNTDNGU1.848773——[13]7BTNTABCODOE1.888060100%3)80%3)[14]8TNPDU1.939034*27.58[20]9HHTDD2.079546100%3)100%3)[45]10TDCD1.99*8410*7.2-11≥2.94)[25]
续表1
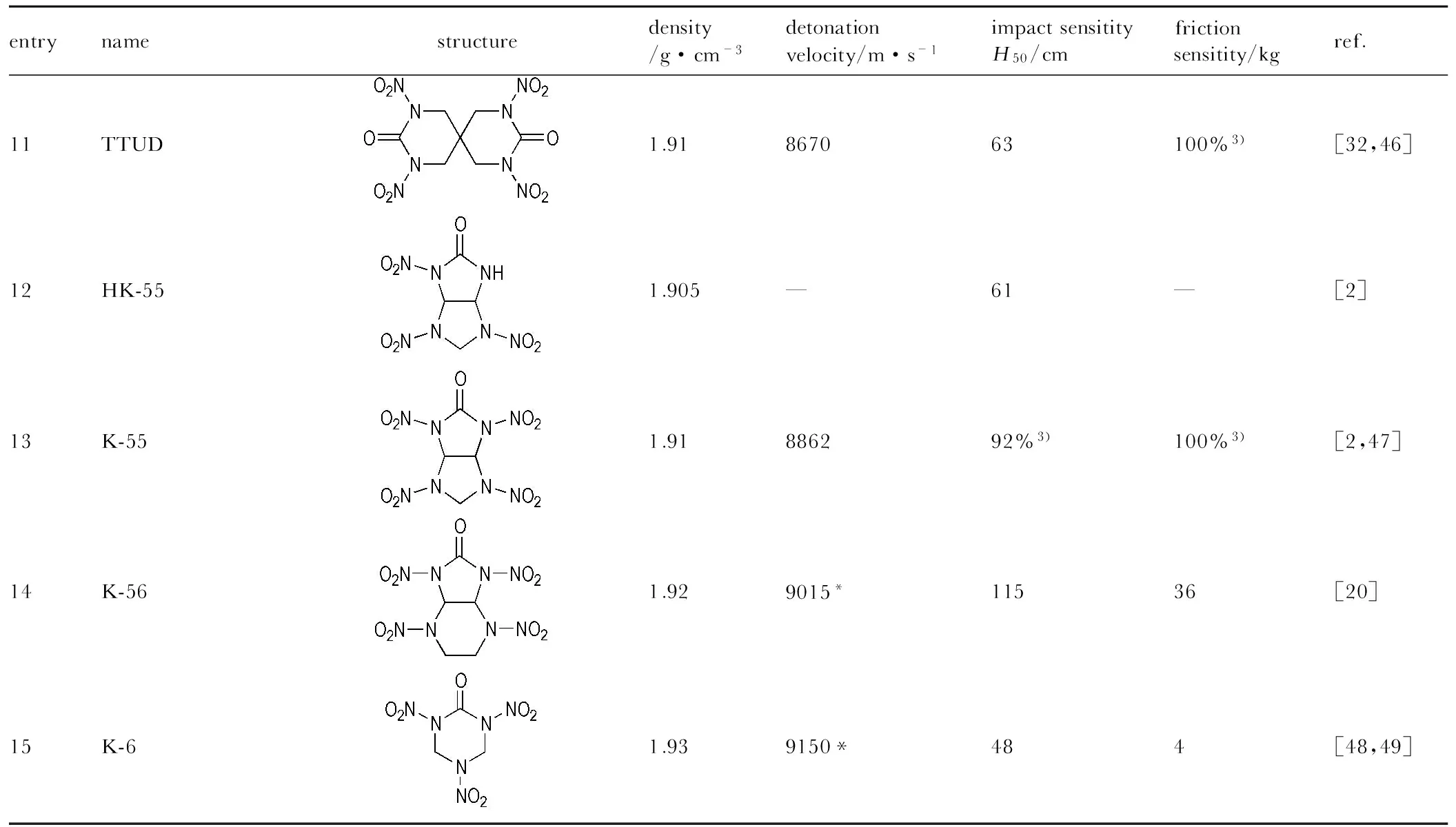
entrynamestructuredensity/g·cm-3detonationvelocity/m·s-1impactsensitityH50/cmfrictionsensitity/kgref.11TTUD1.91867063100%3)[32,46]12HK-551.905—61—[2]13K-551.91886292%3)100%3)[2,47]14K-561.929015*11536[20]15K-61.939150*484[48,49]
Note: 1) All values listed above are measured by experiment, except those marked with an asterisk, which are calculated values; 2) Measured by the Julius Peters equipment, the impact sensitivity is in J and the friction sensitivity is in N; 3) Standard Methods 601.2 and 602.1 of GJB772A-1997; 4) Allegany Ballistics Laboratory test (per MIL-STD-1751), threshold initiation value for 10/10 no-fires, log10(1bf).
由表1可知,与其它系列含能材料相比,硝基脲类含能材料的密度大部分不低于1.90 g·cm-3,其中1,3,4,6-四硝基甘脲(TNGU)和2,4,6,8,10,12-六硝基-2,4,6,8,10,12-六氮杂三环[7.3.0.03.7]十二烷-5,11-二酮(HHTDD) 的密度达到了2.0 g·cm-3。
五元单环硝基脲类含能材料DNDACPO[12]的密度与RDX(1.70 g·cm-3[50])相当,但爆速较低,稳定性差,很难应用到火工品和炸药等含能材料领域中。可通过增加分子中的并环数和致爆基团数目来提高化合物的密度和能量。甘脲类含能化合物TNGU是CHNO类高能含能材料中密度最大的一种炸药,爆炸性能很强[51]。DNGU是单硝基脲类化合物,与TNGU相比,其能量较低[51],被认为是一种钝感高能炸药(IHE),其撞击较为钝感主要归功于分子内部硝脲骨架中的氢键。由于DNGU较为钝感,曾被建议用作不敏感炸药代替RDX和TNT[2]。
TNGU能量高但易水解的特性限制了其应用,甘脲类硝基衍生物TNDGU[4]中的八元环结构具有较高的能量,特有的氮杂环结构增加了其密度和稳定性。其爆轰性能与RDX(爆速约8440 m·s-1[50])相当,感度较低,稳定性和安全性好,且密度稍高于RDX(1.70 g·cm-3[50]),具有较好的综合性能,可用于炸药、推进剂以及火工品等含能材料领域中。甘脲类硝仿基化合物BTNDNG和DMBTNTDNGU[12-13]的密度与TNGU相比略有降低,但爆速高,具有良好的热稳定性和水解稳定性,是一种性能良好的高能材料。化合物BTNTABCODOE[14]的密度和爆速较TNGU有所下降,因该化合物的分子结构环数较多,又有烷撑基存在,引起分子特征密度降低,从而使分子体积变大,导致其密度和爆速均相对较低,这是烷撑双环脲炸药的一个缺陷。但综合分析,化合物BTNTABCODOE是非对称取代的新型环脲硝胺炸药,具有相当高的能量并改善了水解稳定性,可作为火炸药配方的组分。
螺环化合物TTUD[32]能量和密度均较大,撞击感度低,热稳定性良好,综合性能比RDX稍好。六元螺环既可引入致爆基团,又可作为密堆积的骨架分子提高其结晶密度,是研究合成新型硝基脲类含能材料的一个新方向。多环化合物TNPDU[20]和TDCD[25]等均是高性能双硝基脲炸药,密度和爆速均较高。与TNGU相比,TDCD具有很好的化学稳定性、热稳定性和水解稳定性,可应用到炸药和火工品等含能材料领域中。HHTDD是典型的多环氮杂多硝基脲化合物,致爆基团数目的增多,特有的氮杂环结构和环张力的存在使其具有高爆速高密度等良好的爆轰性能,相比之下,其无论密度还是能量均高于其它环脲类化合物[45]。肖鹤鸣等[45]明确指出,HHTDD是目前已知爆速最高的高能化合物,具有深远的研究意义和广阔的应用前景,但摩擦感度和撞击感度也比较高,水解稳定性也较差,这些特点限制了其在炸药上的应用空间。
7 结论与展望
硝基脲类含能材料具有密度大和能量高的特点,部分化合物具备了高能钝感特性,是一种极具研究和应用价值的含能材料。通过对硝基脲类化合物的深入研究,人们相继开发出了诸多合成方法,其中五元环脲类化合物研究得较充分,主要是通过以脲为原料合成,同时辅助以胍类化合物或其它小分子为原料合成。以脲为原料合成法原料便宜易得,反应步骤简单,硝化方法多样,为该类化合物的合成提供了多种途径; 其它辅助方法是对以脲为原料合成法的补充,对于相对较难合成的化合物提供了一条新的途径,该法对于多元环化合物的合成优势更加明显。以—NO2为致爆基团的CHNO类炸药,其晶体密度的最大值为2.2 g·cm-3,其爆热的最大值为7.25 kJ·g-1 [52]。新型CHNO类含能材料仍是近几十年的主要研究内容,硝基脲类含能材料因具有密度大、感度低的特性优势仍具有提高爆速和密度的空间,寻找良好的碳骨架对于高能量密度材料(HEDM)的合成至关重要。
在设计新型硝基脲类化合物时,应尽可能实现硝基脲类化合物的最大应用潜力,可进一步开展下述研究:
(1)目前已报道的甘脲类含能材料具有热稳定性好、能量水平高等优点,但缺乏系统性研究。目前已合成出来的主要有TNGU和TNDGU等,而且TNDGU爆轰性能与RDX相当,具有较好的综合性能。可将甘脲多聚体作为母体进行结构修饰和衍生,完善对甘脲多聚体硝基衍生物的研究。
(2)设计良好的氮杂环骨架分子,引入硝仿基(—C(NO2)3)、硝基(—NO2)、氨基(—NH2)和硝氨基(N—NO2)等含能基团,使化合物具有较好的热稳定性和水解稳定性、较低的感度和较高的爆轰能力。
(3)七元环和八元环硝基脲类化合物研究相对较少,可通过设计、合成新型化合物来完善这一领域,提高硝基脲类化合物的多方面性能并加强其实际应用。
参考文献:
[1] Pagoria P F, Mitchell A R, Jessop E S. Nitroureas II. Synthesis of bicyclic mono- and dinitrourea compounds[J].PropellantsExplosivesPyrotechnics,1996,21: 14-18.
[2] Pagoria P F, Lee G S, Mitchell A R, et al. A review of energetic materials synthesis[J].ThermochimicaActa, 2002, 384: 187-204.
[3] Sikder A K, Sikder N. A review of advanced high performance, insensitive and thermally stable energetic materials emerging for military and space applications[J].JournalofHazardousMaterialsA, 2004, 112: 1-15.
[4] 孟子晖,徐光瑞,徐志斌,等. 四硝基甘脲二聚体、制备方法及应用: CN 102875557[P]. 2013.
MENG Zi-hui, XU Guang-rui, XU Zhi-bin, et al. tetranitrodiglycolurils and its preparation method & application: CN 102875557[P]. 2013.
[5] CUI K J, XU G R, XU Z B, et al. Synthesis and Characterization of a Thermally and Hydrolytically Stable Energetic Material based on N-Nitrourea[J].PropellantsExplosivesPyrotechnics,2010,35: 1-9.
[6] 彭忠吉,万道正. 四硝基甘脲及其水解产物的合成研究[J]. 兵工学报,1980(3): 23-27.
PENG Zhong-ji, WAN Dao-zheng. The synthetic study of tetranitroglycoluril and its hydrolyzed product[J].ActaArmamentarii, 1980 (3): 23-27.
[7] Boileau J, Emeury J L, Kehren J. Tetranitroglycoluril, Herstellungsverfahren dafuer und verwendung als explosivesivstoff: DE2435651[P], 1975-02-06.
[8] Boileau J, Emeury J L, Kehren J P. Tetranitroglycoluril and method of preparation thereof: US 4487938[P], 1984-12-11.
[9] ZHENG Yuan-yang, ZHOU Ji-zhang, ZHANG ming-nan, et al. The synthesis of 1,1,2,2-tetrakis(difluoroaminomethyene- nitroamino)ethane and its related compounds[J].ActaArmamentarii, 1988 (1): 59-63.
[10] 安乔. 甘脲的硝基衍生物的合成研究[D]. 南京理工大学, 2013.
AN Qiao. Synthesis of nitroderivatives of glycoluril[D]. Nanjing University of Science & Technology, 2013.
[11] 易文斌,安乔,蔡春. 一种1,3,4,6-四硝基甘脲的制备方法[P]: CN 103242319A, 2013.
YI Wen-bin, AN Qiao, CAI Chun. A preparation method of 1,3,4,6-tetranitroglycoluril: CN 103242319A[P], 2013.
[12] FANG Yin-gao, WU Guo-hua. Synthesis and properties of 1,4-dinitro-3,6-bis(trinitroethyl)glycoluril[J].ChineseJournalofEnergeticMaterials(HannengCailliao),1997,5(1): 9-14.
[13] ZHAO H A, HU R Z, YANG D S, et al. Kinetics and mechanism of the exothermic first-stage decomposition reaction for 1,5-dimethyl-2,6-bis(2,2,2-trinitroethyl)-4,8-dinitroglycoluril[J].ThermochimicaActa,2004,416: 1-4.
[14] TAN Guo-hong. Synthesis and nitration of some new cyclourea compounds[J].HuaxueShijie,1995,36(10): 530-537.
[15] 任玉杰,刘马. 2-咪唑烷酮衍生物的合成研究进展[J]. 上海应用技术学院学报(自然科学版),2011,11(3): 205-211.
REN Yu-jie, LIU Ma. Review of the synthesis of 2-imidazolidinone derivatives[J].JournalofShanghaiInstituteofTechnology(NaturalScience),2011,11(3): 205-211.
[16] Bachmann W E, Horton W J, Jenner E L, et al. The nitration of derivatives of ethylenediamine[J].JournaloftheAmericanChemicalSociety,1950,72(7): 3132-3134.
[17] Kuchurov I V, Fomenkov I V, Zlotin S G, et al. Nitration of carbonic, sulfuric and oxalic acid-derived amides in liquid carbon dioxide[J].MendeleevCommunications,2013,23(2): 81-83.
[18] Gagnon J L, Zajac W W. Synthesis ofcis-1,5-dimethyl-2,4-dinitro-2,4-diazabicyclo[3.1.0]hexan-3-one andcis-1,5-dimethyl- 2,4-dinitro-2,4-diazabicyclo[3.2.0]heptan-3-one[J].SyntheticCommunications,1996,26(4): 837-845.
[19] Graindorge H R, Lescop P A, Terrier F, et al. Synthesis of 2,5,7,9-tetranitro-2,5,7,9-tetraazabicyclo[4.3.0]nonanone[C]∥Proceedings of the 211st American Chemical Society National Meeting, Washington, DC, 1996, 623: 43-50.
[20] Sikder A K, Bhokare G M, Agrawal, Sarwade D B, et al. Synthesis, characterization and thermal behaviour of 2,4,6,8-tetranitro- 2,4,6,8-tetraazabicyclo[3.3.1]nonane-3,7-dione(TNPDU) and one of its methylene analogues[J].PropellantsExplosivesPyrotechnics,2001,26: 63-68.
[21] Adolph H G, Boyer J H, Dagley I J, Flippen J L, et al. Facile synthesis and nitration of cis-syn-cis-2,6-dioxodecahydro- 1H,5H- diimidazo[4,5-b:4′,5′-e]pyrazine[J].TheJournalofOrganicChemistry,1991,56(10): 3413-3419.
[22] Vedachalam M, Boyer J H. 2,6-Dithiodecahydro-1H,5H-Diimidazo[4,5,-b:4′,5′-e]Pyrazine and related dioxo- and diimino- decahydrodiimidazopyrazines[J].HeteroatomChemistry,1993,4(1): 85-90.
[23] McKay A F, Wright G F. The nitration products of 2-nitramino-△2-1,3-diazacycloalkenes[J].JournaloftheAmericanChemicalSociety,1948,70(12): 3990-3994.
[24] Boyer J H, Ramakrishnan V T, Vedachalam M, Dense compounds of C, H, N, and O atoms: Nitramine derivatives of diimino- and dioxodecahydro-1H,5H-diimidazo-[4,5-b:4 ′,5′ -e]pyrazine[J].HeteroatomChemistry,1991,2(2): 313-318.
[25] Fischer J W, Hollins R A, Lowe-Ma C K, et al. Synthesis and characterization of 1,2,3,4-cyclobutanetranitramine derivatives[J]. theJournalofOrganicChemistry,1996,61: 9340-9343.
[26] Mitchell A R, Pagoria P F, Coon C L, et al. NitroureasⅠ. Synthesis, scale-up and characterization of K-6[J].PropellantsExplosivesPyrotechnics,1994,19(5): 232-239.
[27] 周诚,周彦水,霍欢. 1,3,5-三硝基-六氢化-1,3,5-三嗪-2-酮的合成与表征[J]. 火炸药学报,2011,34(4): 17-20.
ZHOU Cheng, ZHOU Yan-shui, HUO Huan. Synthesis and characterization of 1,3,5-trinitro-hexahydro-1,3,5-triazin-2(1H)-one[J].ChineseJournalofExplosives&Propellants,2011,34(4): 17-20.
[28] McKay A F, Manchester D F. The nitration products of some substituted 2-nitramino-1,3-diazacycloalkene-2[J].JournalofAmericanChemicalSociety,1949,71(6): 1970-1973.
[29] Huang D S, Rindone R R. High-energy insensitive cyclic nitramines: US 4983734[P]. 1991.
[30] Huang D S, Rindone R R. Preparationof 2,4,6-trinitro-2,4,6-triazacyclohexanone: US 5391736[P]. 1995.
[31] 周诚,周彦水,王伯周,等. 1.3.5-三硝基-1,3,5-三嗪-2(1H)-酮(Keto-RDX)新法合成、晶体结构和热性能研究[J]. 有机化学,2012,32: 75-80.
ZHOU Cheng, ZHOU Yan-shui, WANG Bo-zhou, et al. A novel synthetic route, crystal structure and thermal behaviors for 1,3,5- trinitro-hexahydro-1,3,5-triazin-2(1H)-one (Keto-RDX)[J].JournalofOrganicChemistry,2012,32: 75-80.
[32] FU Quan-jun, ZHU Chun-hua, FU Xia-yun. On the synthesis, structure and properties of spiro-nitramines and spiro-nitrosamines[J].ActaArmamentaria,1992,1: 28-35.
[33] Oxley J C, Smith J L, Vadlamannati S. Synthesis and characterization of urea nitrate and nitrourea[J].PropellantsExplosivesPyrotechnics,2013,38: 335-344.
[34] Oxley J L, Smith J L, Naik S, et al. Decompositions of urea and guanidine nitrates[J].JournalofEnergeticMaterials,2009,27: 17-39.
[35] 杨建明,余秦伟,薛云娜,等. 二硝基脲的合成、表征及热力学的理论研究[J]. 含能材料,2011,19(2): 160-164.
YANG Jian-ming, YU Qin-wei, XUE Yun-na, et al. Synthesis, characterization and theoretical research of thermodynamics on dinitrourea[J].ChineseJournalofEnergeticMaterials(HannengCailliao), 2011,19(2): 160-164.
[36] Patrick G, Niklas W, Helena B, Synthesis and analyses of N,N′-dinitrourea[J].PropellantsExplosivesPyrotechnics,2001,26: 17-20.
[37] 李亚妮,杨建明,余秦伟,等. 二硝基脲含能离子盐的合成及表征[J]. 含能材料,2011,19(3): 247-253.
LI Ya-ni, Yang Jian-ming, YU Qin-wei, et al. Preparation and characterization of energetic ionic salts of N,N′-dinitrourea,ChineseJournalofEnergeticMaterials(HannengCailliao), 2011,19(3): 247-253.
[38] XI Yu-ye, CAI Zheng-qian, WANG Nai-yan, et al. Relationships between properties and electronic structure of cyclourea nitro-amine compounds Ⅳ. Study of thermodecomposition[J].ChineseJournalofAnalyticalChemistry,1991,19(12): 1387-1391.
[39] Hu R Z, Yang D S, Zhao H A, et al. Kinetics and mechanism of the exothermic first-stage decomposition reaction for 1,4-dinitro-3,6-bis(trinitroethyl) glycoluril[J].ThermochimicaActa,2002,389(1-2): 65-69.
[40] HU Rong-zu, LIANG Yan-jun, FANG Yin-gao,et al. The hydrolytic stability of some cyclourea compounds[J].JournalofThermalAanalysis,1996,46(5): 1283-1289.
[41] 马从明,刘祖亮,姚其正. 环脲硝胺类含能化合物的合成及性能研究进展[J], 含能材料,2014,22(2): 263-269.
MA Cong-ming, LIU Zu-liang, YAO Qi-zheng. Progress in synthesis and performance of cyclourea nitro-amine energetic compounds[J].ChineseJournalofEnergeticMaterials(HannengCailliao), 2014,22(2): 263-269.
[42] 胡荣祖,方银高,卢兴森. 环脲硝胺化合物稳定性的研究[J]. 火炸药,1981(5): 1-11.
[43] Khire V H, Talawar M B, Prabhakaran K V, et al. Spectro-thermal decomposition study of 1,4-dinitroglycoluril (DINGU)[J].JournalofHazardousMaterialsA, 2005,119: 63-68.
[44] 方银高. 二硝基甘脲合成方法的改进[J]. 火炸药,1983(1): 187-204.
[45] 邱玲,肖鹤鸣,居学海,等. 六硝基六氮杂三环十二烷的结构和性能: HEDM分子设计[J]. 化学物理学报,2005,18(4): 541-546.
QIU Ling, XIAO He-ming, JU Xue-hai, et al. Structure and properties of hexanitrohexaazatricyclododecane: molecular design of HEDM[J].ChineseJournalofChemicalPhysics,2005,18(4): 541-546.
[46] HU Rong-zu, ZHAO Feng-qi, WANG Bo-zhou, et al. Kinetic study on the exothermic first-stage decomposition reaction of 2,4,8,10-tetranitro-2,4,8,10-tetraazaspiro[5.5]undecane-3,9-dione[J].ChineseJournalofChemistry,2004,22(10): 1075-1077.
[47] 易景缎. 四硝基半甘脲的合成及其性能[J]. 火炸药,1992(2): 6-10.
[48] Sikder N, Bulakh N R, Sikder A K, et al. Synthesis, characterization and thermal studies of 2-oxo-1,3,5-trinitro-1,3,5- riazacyclohexane (Keto-RDX or K-6)[J].JournalofHazardousMaterialsA,2003,96: 109-119.
[49] Shokrollahi A, Zali A, Pouretedal H R, et al. Synthesis of Keto-RDX and its characterizations calculation[J].ChineseJournalofEnergeticMaterials(HannengCailiao)),2008,16(1): 44-48.
[50] Dodratz B M. Insensitive high explosive triaminotrinitrobenzene(TATB): Development and characterization - 1888 to 1994[R]. LA-13014-H: 1995.
[51] Fedoroff B T, Aaronson H A, Clift G D, et al. Encyclopaedia of Explosives and Related Items[M]. Dover, New Jersey: Picatinny Arsenal,1960, 1, A65.
[52] 董海山. 高能量密度材料的发展及对策[J]. 含能材料,2004(增刊): 1-12.
DONG Hai-shan. The development and countermeasure of high energy density materials[J].ChineseJournalofEnergeticMaterials(HannengCailiao),2004(Suppl.): 1-12.
猜你喜欢
分子催化(2022年1期)2022-11-02
化工管理(2021年7期)2021-05-13
世界农药(2019年4期)2019-12-30
中成药(2018年12期)2018-12-29
中南大学学报(自然科学版)(2016年2期)2017-01-19
中国农资(2016年1期)2016-12-01
合成化学(2015年2期)2016-01-17
合成化学(2015年9期)2016-01-17
天然产物研究与开发(2014年7期)2014-04-27
火炸药学报(2014年5期)2014-03-20
