
分子模拟中常用的结构分析与表征方法综述
2015-05-14 07:19张世良冯士东1刘日平燕山大学理学院河北秦皇岛066004燕山大学亚稳材料制备技术与科学国家重点实验室河北秦皇岛066004
燕山大学学报 2015年3期
张世良,戚 力,高 伟,冯士东1,,刘日平(1.燕山大学理学院,河北秦皇岛066004;.燕山大学亚稳材料制备技术与科学国家重点实验室,河北秦皇岛066004)
分子模拟中常用的结构分析与表征方法综述
张世良1,2,∗,戚 力2,高 伟2,冯士东1,2,刘日平2
(1.燕山大学理学院,河北秦皇岛066004;
2.燕山大学亚稳材料制备技术与科学国家重点实验室,河北秦皇岛066004)
摘 要:分子模拟技术是随着计算机技术发展而兴起的一项新技术,主要包括分子动力学和蒙特卡洛等方法。通过分子模拟技术,利用计算机对材料的组成、性质、制备工艺、加工工艺等环节进行虚拟实验,获得大量数据,再与验证性实验进行对照,可以分析其中的物理现象和本质。本文整理了目前分子模拟技术中常用的几种结构分析与表征方法,对其原理和分析要点进行综述,希望能够有利于分子模拟技术在国内的推广和应用。
关键词:分子模拟;分子动力学;蒙特卡洛;结构分析;综述
0 引言
分子模拟技术是随着计算机技术发展而兴起的一项新技术,主要包括分子动力学(Molecular dynamics simulation,MD)和蒙特卡洛(Monte Carlo,MC)等方法。通过分子模拟技术,人们可以在分子、原子尺度上对所关心材料的物理、化学性质以及与之相对应的各种现象进行模拟,摒弃传统材料科学研究中被称之为“爱迪生方式”的炒菜式研究,利用计算机对材料的组成、性质、制备工艺、加工工艺等环节进行虚拟实验,获得大量数据,再与少量验证性实验进行对照,分析物理现象和本质,从而大大的降低材料的研发成本和时间。从20世纪90年代起,这种新材料的设计模式已经很大程度上取代了传统的新材料设计方式,成为新材料设计的主要方式之一。2011年,美国总统奥巴马宣布启动一项价值超过5亿美元的“先进制造业伙伴关系”(Advanced Manufacturing Part⁃nership,AMP)计划,而以材料计算与模拟为主的“材料基因组计划”(Materials Genome Initiative,MGI)则是AMP计划的重要内容之一[1]。2012年,《材料科学系统工程发展战略研究——中国版材料基因组计划》重大项目启动会在中国工程院召开,代表着中国版的MGI计划开始实施[2]。可以预见,分子模拟技术在新材料设计以及凝聚态物理等研究领域会越来越发挥出重要作用。
对于分子模拟的结果,可以输出的参数有粒子坐标、粒子速度、粒子受力等等,通过对数据进行分析处理,可以得到动力学特征、热力学特征以及结构特征等信息,而结构分析又是可以与实验研究相对应的参量,因此在模拟研究中更显得尤为重要。
目前,对分子模拟结果结构分析的方法很多,更多的是分散在英文文献中,由于翻译和理解上的差异,有些名词开始出现偏差,影响了国内从事分子模拟科研工作者的理解与交流。本文整理了目前分子模拟技术中常用的结构分析方法,对各种方法的原理和分析要点进行综述,希望能够有利于分子模拟技术在国内的推广和应用。
1 常用的宏观统计分析方法
对分布函数、径向分布函数和静态结构因子都是表征体系结构的宏观统计参量,三者之间可以进行数值的相互转化。由于静态结构因子也可以通过X衍射或中子衍射在实验中得到,因此对分布函数、径向分布函数和静态结构因子这3种方法就可以作为连接计算机模拟与实验分析的桥梁,成为分子动力学模拟中最常使用的结构分析方法。
1.1对分布函数
对分布函数(Pair distribution function,PDF)反映的是从一个任意指定的“中心”粒子出发,到半径为r的位置上出现其它粒子的几率(单位体积内的粒子数目)[3]。如图1所示,取一原子为观测中心,以1 Å为单位向外画出一层层的同心球壳,然后统计每一壳层里的原子数密度(粒子数n(r)/体积V)与平均数密度(ρ0=总粒子数/体积)的比值,即为对分布函数,常用g(r)表示。在国外的文献中,也有称之为对关联函数(Pair Correlation Function,PCF)[4⁃5],在国内一些文献上,也有时将其称双体分布函数、偶分布函数或等,表达式为[6]

其中,r为距中心原子半径,δr为球壳厚度,n(r)为球壳内的粒子数,ρ0为理想晶体中数密度。

图1 对分布函数与原子结构关系的示意图Fig.1 Diagram of the relationship between the PDF and the atomic structure
对于g(r)的分析,主要是基于以下几点:
1)峰的分布规律。根据对分布函数的物理含义,对于晶体,由于原子的周期性排列,每层原子会对应出现分布峰值,而在层与层的原子之间出现原子的几率为0。对于液体结构,由于短程有序,在第一近邻处会出现较为尖锐的峰值,在中远程的范围里,液体结构的对分布函数会出现漫高峰,并且趋近于1。这说明在液态结构中,相对于一个中心原子,在无穷远处会总会有一个原子的存在。面心立方晶体铜和3 000 K温度下液态铜的对分布函数如图2所示。

图2 面心立方晶体铜和3 000 K温度下液态铜的对分布函数比较Fig.2 Comparison of the PDF between the copper with face⁃centered cubic(FCC)structure and the copper with liquid structure at 3 000 K
2)第一峰的高度和形状。第一峰代表第一近邻原子之间的结合强度,如果它的外形比较尖锐,表明在此半径范围内的原子数密度比平均密度要高很多,中心原子与最近邻原子的相互结合强度也比较大。
3)第一峰与第一谷的关系。Abraham[7]等人提出一种经验性方法,根据对分布函数中第一谷值gmin与第一峰值gmax的比例,定义参量R=gmin/gmax,用来确定玻璃转变点Tg,如图3(a)所示。
4)第二峰的形状与分布。第二峰代表着中程序的连接强度,如果第一峰和第二峰比较明显,同时第一峰与第二峰之间有一个很深的波谷,基本上表明原子是以一种共价键的形式存在。如果第二峰出现了劈裂,则这是出现非晶结构的典型特征[8⁃9]。

图3 PCF参量、原子体积与温度关系Fig.3 Relationship of the Abraham parameter R,atomic volume and temperature
1.2径向分布函数
径向分布函数(Radial Distribution Function,RDF)表示在半径为r处厚度δr的球壳内的平均原子数,其数值等于该位置出现粒子的几率g(r)与球壳面积之积。常用符号G(r)表示,公式为[10]

由于语言转换和理解上的差异,径向分布函数与对分布函数在中外文的文献中经常被互用,有时使用的是径向分布函数G(r)概念,表达出来的却是对分布函数g(r)含义。其实二者无论是在图像上还是物理意义上都有明确的区别[5,11⁃12]。如图4所示,对分布函数g(r)表示的是概率,对于液体或非晶结构,分布函数曲线随着半径r的增加应该是归一的,如图4(a);而径向分布函数G(r)则表示在球面上粒子的统计值,曲线随着半径的增加整体上升,如图4(b)。从目前的发展趋势来看,多数学者都已认同了用径向分布函数的名称来表示对分布函数的用法。
对于二元或多元体系,根据不同类型原子之间的相互分布情况,还可以用偏对关联函数(Partial Pair Correlation Function,PPCF)来表征在多元体系中不同原子的排列规律[13⁃14]。图5所示为Cu64Zr36合金快速至700 K时的偏对关联函数曲线。从图中可以看出,总对关联函数是由不同原子之间(Zr⁃Zr,Cu⁃Zr,Cu⁃Cu)的相互分布曲线加权迭加而成。其中,由于Cu的原子半径要小于Zr的原子半径,因此以Cu为中心原子,其它Cu原子为近邻原子时,Cu⁃Cu之间的距离r较小,而以Cu为中心原子,Zr原子为近邻原子时的几率最高,也就是说,Cu更倾向于与Zr形成近邻。
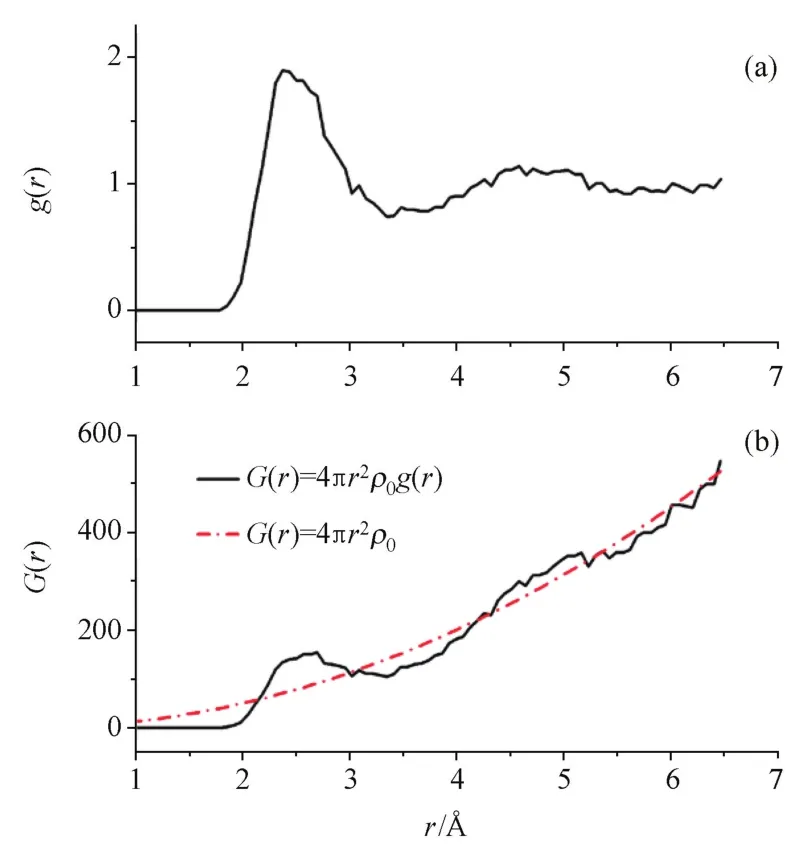
图4 对分布函数与径向分布函数的关系Fig.4 Relationship between PDF and RDF

图5 700 K温度下Cu64Zr36合金的偏对关联函数Fig.5 PPCF curve of Cu64Zr36 alloy at 700 K
1.3静态结构因子


静态结构因子(S(k)-1)与对分布函数(g(r)-1)互为傅里叶变换,在粒子的空间结构信息中,静态结构因子为倒易空间,对分布函数为实空间。二者之间的三维傅里叶变换式[3]为

实际上,g(r)函数是一个球对称的函数,可以将三维的变量转为仅与半径尺度有关的一维变量,因此式(4)转换为半径积分的一维傅里叶变换,静态结构因子与对分布函数的关系式可以表示为[3]

经过傅立叶变换后的对分布函数与静态结构因子曲线如图6所示。对于结构因子的分析,基本上也是针对峰的位置、峰的形状进行判断。

图6 300 K温度下Cu非晶的g(r)与S(k)形状对比Fig.6 Comparison of the curve between the g(r)and S(k)of glassy copper at 300 K
1)峰位置的相对关系。由于结构因子是倒易空间,变量k与距中心原子的半径r成反比关系。根据第二峰位与第一峰位k2/k1的比值,可以将液体分为3种类型。第一种为大部分的液态金属,比值为1.86;第二种为Ga、Sn、Bi等液体,比值为1.96,其特征为在第一峰右边存在一个肩峰;第三种为In和Tl,比值为1.88[10];
2)预峰的特征。在第一峰的前面,如果出现小的预峰,反映的是体系中存在着中程序结构特征[10];
3)熔点温度峰值特征。通过对多种液体的统计发现,液态结构因子的第一峰高度在熔点附近都会达到一个同样的高度,即S(k)≈2.8,这个规律叫做Hansen⁃Verlet凝固判据,是可以用来作为熔化和凝固的判据之一[3]。
2 常用的几种共近邻分析方法
共近邻分析(Common neighbor analysis,CNA)是用来描述一个原子周围近邻原子的排列特征的方法,根据计算方法的不同,可包括键对分析、键角分析、泰森多边形指数分析、配位数等。最近邻的距离一般是采用对分布函数的第一低谷处的原子位置值rmin。
2.1键对分析
键对分析(Pair analysis,PA)是研究液体和固体中团簇的一种常用的有效方法[16],也有文献将这种方法用创始人的名字命名为Honeycutt⁃Andersen指数(HA index)[17]。这种方法用4个指数i、j、l和m对原子进行分类标定,用1551、1541等指数形式来表征体系中存在的结构特征。i=1表示研究的两个原子对能够成键,i=2表示两个原子对不能成键,一般我们都关注于成键原子(i=1)之间的关系。j表示两个被研究原子对中相同的近邻原子数。l则表示j个相同的近邻原子中成键的原子对数。m表示l中成键近邻原子的排布情况,如果成键相连,则m为1,否则m为2[18]。
根据上述定义规则,在面心立方(FCC)和密排六方(HCP)结构中以1421为主,体心立方(BCC)结构中以1441和1661为主。总体上来看,在液体和非晶结构中大量存在着1551、1541、和1431结构。其中1551键对表征完整的二十面体(图7所示),1541和1431则表征缺陷二十面体[19]。
2.2键角分布
键角分布(Bond angle distribution,BAD)就是体系中所有原子与其它最近邻原子成键所构成的键角的分布,所获取的是原子在三维空间上的排布信息,因而可以弥补对分布函数g(r)仅仅依据原子距离结构获取信息的不足[8]。根据几何算法,对于标准的多面体,键角的分布情况应该如表1所示。然而由于温度的作用,原子之间的振动会影响实际的角度分布情况,因此从角度的分布变化上,可以看出体系中的二十面体或四面体的扭曲和破坏情况[20⁃22]。图8所示为3 000 K温度下Cu熔液的角分布函数。从角分布两个高峰值来看θ1=55°,θ2=104°,熔液中应该存在着二十面体或四面体,但角度均小于标准二十面体的θ1=63.4°、θ2=116.6°和理想四面体的θ1=109.5°,这表明液态结构配位原子之间的距离要长于晶态时原子之间的距离,导致成键原子之间的夹角偏小。另外,更多的缺陷二十面体的存在,也致使液态结构的角度与标准结构的角度分布出现偏差。

图7 键对1551与二十面体关系示意图Fig.7 Diagram of the relationship between the HA index 1551 and the icosahedron
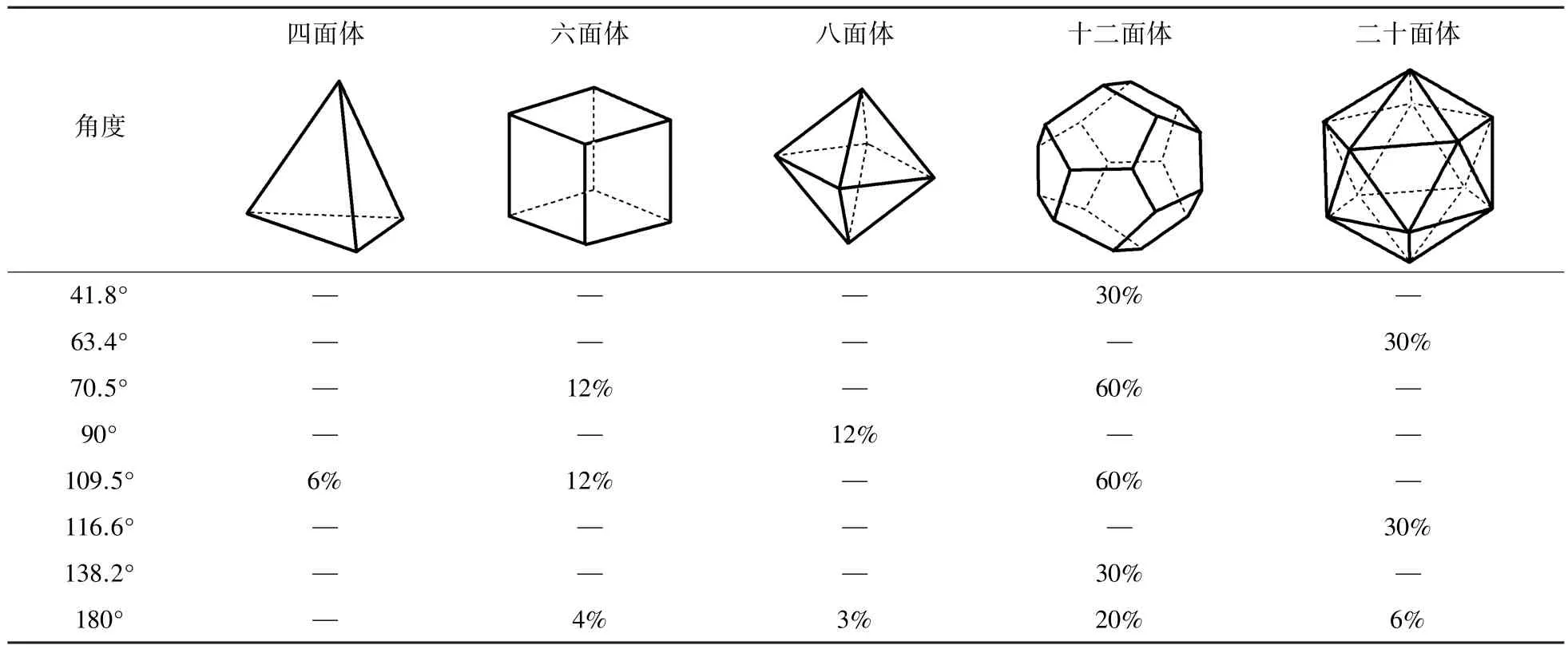
表1 标准多面体的键角分布Tab.1 Bond angle distribution of standard polyhedron

图8 3 000 K温度下Cu熔液的角分布函数Fig.8 Bond angle distribution of liquid copper at 3 000 K
2.3泰森多边形指数分析
泰森多边形(Voronoi polyhedral,VP)是一种几何算法,在气象、物流、商业等多个行业和领域中已被广泛利用,也常被称为Voronoi网格、Voronoi划分或Dirichlet图。它是由两个邻点连接直线的垂直平分线组成的连续多边形组成,二维泰森多边形原理示意图如图9所示。这种方法可以描述在平面上的N个点,按照最邻近原则划分与它的最近邻区域的相互关系。利用Voronoi算法,将离散的原子进行三维空间上的划分,利用所得到的空间结构来表征材料的微观结构,目前也已成为结构表征的一种常用手段。泰森多边形指数常用<n3,n4,n5,n6>的形式表示,其中ni代表着Voronoi多面体具有的i边形数[21,23⁃24]。例如:<0,4,0,0>是由4个四边形所组成的四面体;<0,6,0,0>是由6个四边形所组成的六面体;<2,3,0,0>是由2个三边形和3个四边形所组成的五面体;<0,3,6,0>是由3个四边形和6个五边形所组成的九面体。<0,0,12,0>表示由12个五边形所组成的二十面体。标准多面体的Voronoi指数在表2中列出。晶胞中的Voronoi多面体花纹和指数为<0,3,6,0>的结构示意图如图10所示[8]。
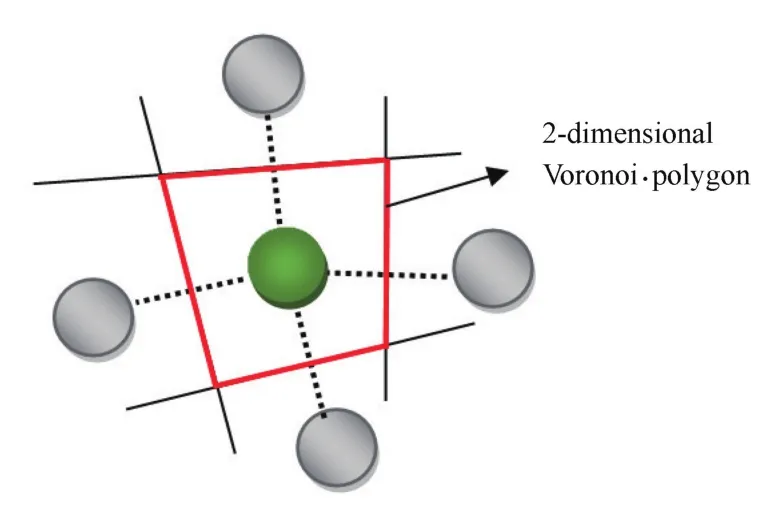
图9 二维Voronoi多边形原理示意图Fig.9 Schematic diagram of 2D Voronoi polygon
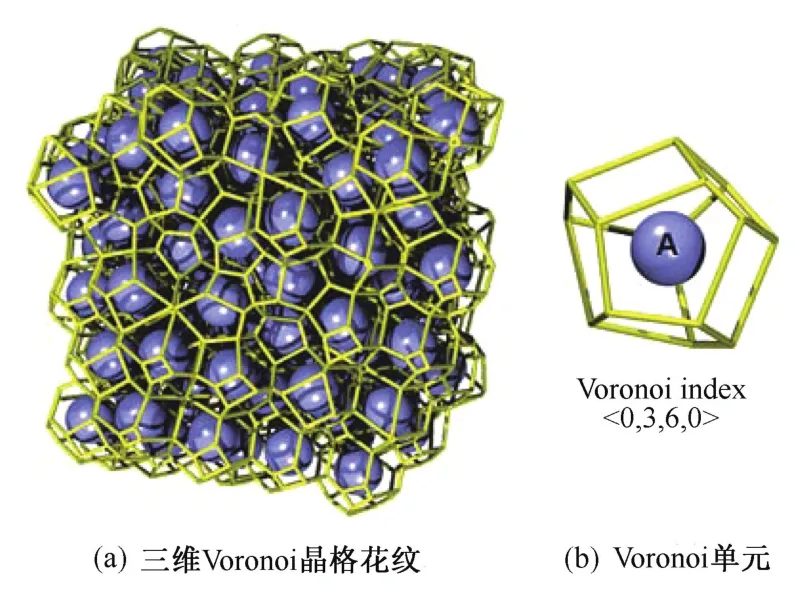
图10 Voronoi多面体示意图[8]Fig.10 Diagram of Voronoi polygon
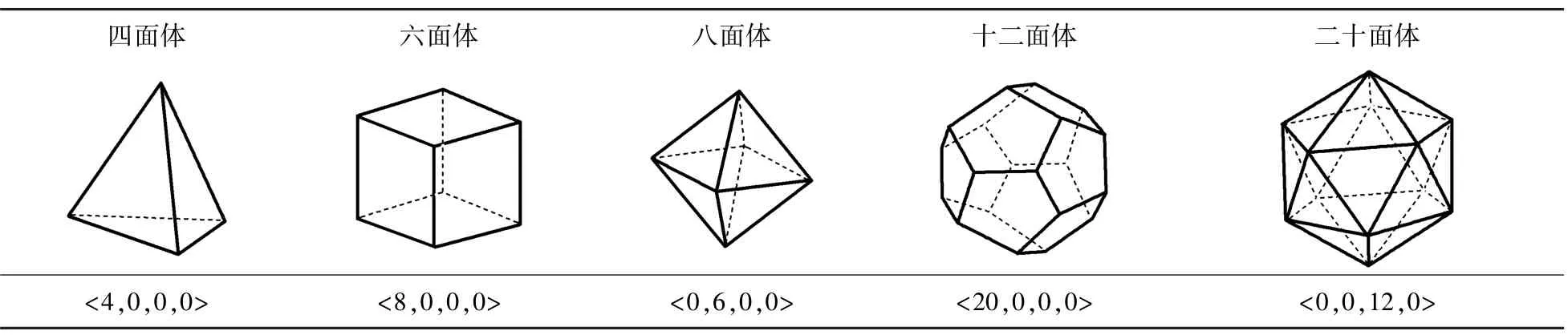
表2 标准多面体的Voronoi指数Tab.2 Voronoi index of standard polyhedron
2.4配位数
第一近邻的配位数(Coordination number,CN)也是一种传统的分析微观结构的方式,此概念首先由阿尔弗雷德·维尔纳在1893年提出,原来更多地用于化学结构的表征,是配位化学的基础。在微观结构的表征中,配位数是用来描述中心原子第一壳层内原子的平均数目,反映的是中心原子与其它原子的结合能力和配位关系,描述的是体系中粒子排列的紧密程度,配位数越大,粒子排列越紧密。一般来说配位数是通过对径向分布函数的第一峰进行积分来得到,其定义为

对于Voronoi指数分析而言,由于泰森多边形的算法就是计算中心原子与配位原子之间的连线平分面,因此,通过对Voronoi指数的累加,也能得到中心原子的配位数。
对于晶体结构,通过配位数可以判断出晶体的结构;对于液态的非晶态结构,配位数可以作为一个发生结构转变的敏感参量,为结构转变的判断提供依据。例如,对于液体中发生的液⁃液相变现象,众多研究[25⁃28]都将配位数的变化作为判断的依据之一。标准堆积模型的配位数与致密度(Relative denisty)、原子间最近距离(Nearest⁃dis⁃tance)相关,如表3所示。

表3 标准堆积模型的配位数Tab.3 Coordination number of standard close⁃packed model
3 展望
文中所综述的结构分析表征方法,基本上还是以短程序(Short⁃range order,SRO)的结构分析为主,其主要标志就是统计和表征第一近邻内的原子结构信息。因此,势函数的截断值(cutoff)和第一近邻的截断值rmin的选取会成为影响分析的重要因素。另外,对分布函数等宏观统计信息很难表现出局部具体特征,而共近邻分析等方法所表现出来的局部特征有时又不具有统计性,很难代表体系的总体特征。因此,通过多种表征方式对分析的结构互相验证是进行分子模拟中结构分析时避免误差的有效方法之一。
随着中程序(Medium⁃range order,MOR)的提出和发展[14,17,29],对团簇的中程序研究,甚至是长程序的表征已成为总体趋势。例如,根据第一近邻和第二近邻中间的rdef值,根据与标准面心立方的配位数的关系而定义的缺陷原子数Ndef方法[30]、结构有序参数[31]等缺陷表征方法;用来表征团簇中程序结构连接的团簇类型指数(Cluster⁃type Index Method,CTIM)方法[32];根据Voronoi多面体互为最近邻原子的原子对数而定义的团簇相关性方法[24]等。无论是中程序,还是长程序,其本质都是在寻求短程序范围内的团簇之间结合方式。因而,对短程序的结构分析则是分子模拟中结构分析与表征的基础,对材料的分子模拟研究至关重要。
参考文献
[1]赵继成.材料基因组计划简介[J].自然杂志,2014,36(2):89⁃104.
[2]杨炳忻.香山科学会议第415⁃419和S14次学术讨论会简述[J].中国基础科学,2012(3):35⁃42.
[3]March N H,Tosi M P.Introduction to liquid state physics[M].Singapore:World Scientific Publishing Co.Pte.Ltd,2002:75⁃80
[4]Jakse N,Pasturel A.Dynamic aspects of the liquid⁃liquid phase transformation in silicon[J].The Journal of Chemical Physics,2008,129(10):104503.
[5]Kim T H,Kelton K F.Structural study of supercooled liquid transi⁃tion metals[J].The Journal of Chemical Physics,2007,126 (5):054513.
[6]陈正隆,徐为人,汤立达.分子模拟的理论与实践[M].北京:化学工业出版社,2007:110⁃112.
[7]Abraham F F.An isothermal⁃isobaric computer simulation of the supercooled⁃liquid/glass transition region:Is the short⁃range order in the amorphous solid fcc?[J].The Journal of Chemical Physics,1980,72(1):359⁃365.
[8]Cheng Y Q,Ma E.Atomic⁃level structure and structure⁃property relationship in metallic glasses[J].Progress in Materials Science,2011,56(4):379⁃473.
[9]Qi L,Domg L F,Zhang S L,et al.Glass formation and local struc⁃ture evolution in rapidly cooled PdNi alloy melt under high pressure[J].Physics Letters A,2008,372(5):708⁃711.
[10]孙民华,牛丽.液态物理概论[M].北京:科学出版社,2013:3⁃20.
[11]Laio A,Vandevondele J,Rothlisberger U.A Hamiltonian electro⁃static coupling scheme for hybrid Car⁃Parrinello molecular dy⁃namics simulations[J].The Journal of Chemical Physics,2002,116(16):6941⁃6947.
[12]Kresse G,Hafner J.Ab initio molecular dynamics for liquid metals [J].Physical Review B,1993,47(1):558⁃561.
[13]Peng H L,Li M Z,Wang W H.Structural signature of plastic de⁃formation in metallic glasses[J].Physical Review Letters,2011,106(13):135503.
[14]Sheng H W,Luo W K,Alamgir F M,et al.Atomic packing and short⁃to⁃medium⁃range order in metallic glasses[J].Nature,2006,439(7075):419⁃425.
[15]赵刚.液态合金微观结构的分子动力学模拟[D].合肥:中国科学院合肥物质科学研究院,2007.
[16]Qi L,Dong L F,Zhang S L,et al.Glass formation and local struc⁃ture evolution in rapidly cooled Pd55Ni45 alloy melt:Molecular dynamics simulation[J].Computational Materials Science,2008,42(4):713⁃716.
[17]Kim T H,Goldman A I,Kelton K F.Structural study of supercooled liquid silicon[J].Philosophical Magazine,2008,88 (2):171⁃179.
[18]Honeycutt J,Andersen H.Molecular dynamics study of melting and freezing of small Lennard⁃Jones clusters[J].Journal of Phys⁃ical Chemistry,1987,91(19):4950⁃4963.
[19]胡壮麒,王鲁红,刘轶.电子和原子层次行为的计算机模拟[J].材料研究学报,1998,12(1):1⁃8.
[20]Liu C S,Zhu Z G,Xia J,et al.Molecular dynamics simulation of the local inherent structure of liquid silicon at different tempera⁃tures[J].Physical Review B,1999,60(5):3194⁃3199.
[21]Zhang S L,Zhang X Y,Wang L M,et al.Voronoi structural evolu⁃tion of bulk silicon upon melting[J].Chinese Physics Letters,2011,28(6):067104.
[22]Iishimaru M,Yoshida K,Kumamoto T,et al.Molecular⁃dynamics study on atomistic structures of liquid silicon[J].Physical Review B,1996,54(7):4638⁃4641.
[23]Finney J L.Random packings and the structure of simple liquid.I.The geometry of random close packing[J].Proceedings of the Royal Society of London,Series A,1970,319(1539):479⁃493.
[24]Li M,Wang C,Hao S,et al.Structural heterogeneity and medium⁃range order in ZrxCu100⁃x metallic glasses[J].Physical Review B,2009,80(18):1⁃7.
[25]Sastry S,Austen Angell C.Liquid⁃liquid phase transition in super⁃cooled silicon[J].Nature Materials,2003,2(11):739⁃743.
[26]Jakse N,Pasturel A.Liquid⁃liquid phase transformation in silicon:evidence from first⁃principles molecular dynamics simula⁃tions[J].Physical Review Letters,2007,99(20):205702.
[27]Vasisht V V,Saw S,Sastry S.Liquid⁃liquid critical point in super⁃cooled silicon[J].Nature Physics,2011,7(7):549⁃553.
[28]Zhang S L,Wang L M,Zhang X,et al.Polymorphism in glassy sil⁃icon:Inherited from liquid⁃liquid phase transition in supercooled liquid[J].Scientific Reports,2015,5:8590.
[29]Gladden L F,Elliott S R.Computer⁃generated models of a⁃SiSe2:Evidence for a glass exhibiting medium⁃range order[J].Physical Review Letters,1987,59(8):908⁃911.
[30]Lutsko J,Wolf D,Phillpot S.Molecular⁃dynamics study of lattice⁃defect⁃nucleated melting in metals using an embedded⁃atom⁃ method potential[J].Physical Review B,1989,40(5):2841⁃2855.
[31]Agrawal P M,Rice B M,Thompson D L.Molecular dynamics study of the melting of nitromethane[J].The Journal of Chemical Physics,2003,119(18):9617⁃9627.
[32]Liu R,Dong K,Tian Z,et al.Formation and magic number char⁃acteristics of clusters formed during solidification processes[J].Journal of Physics:Condensed Matter,2007,19(19):196103.
Summary of methods for structural analysis and characterization in molecular modeling
ZHANG Shi⁃liang1,2,QI Li2,GAO Wei2,FENG Shi⁃dong1,2,LIU Ri⁃ping2
(1.School of Sciences,Yanshan University,Qinhuangdao,Hebei 066004,China;2.State Key Laboratory of Metastable Materials Science and Technology,Yanshan University,Qinhuangdao,Hebei 066004,China)
Abstract:Molecular modeling is developed as the computering aibility is greatly enhanced,and it mainly includes the molecular dy⁃namics simulation and Monte Carlo.Molecular modeling simulation enables the determination of a large number of data regarding materials′composition,properties,preparation and processing by virtual experiments on computers.Compared with the experiment esults,the physical nature behind the phenomena can be understood.This paper summarized some methods on the structural analy⁃es and characterization in molecular modeling,and reviewed the basic principles and applications.This work is expected to promote he applications of molecular modeling.
Key words:molecular modeling;molecular dynamics simulation;Monte Carlo;structural analysis;summary
作者简介:∗张世良(1973⁃),男,辽宁北票人,博士,副研究员,主要研究方向为非晶与液态结构的分子动力学模拟,Email:slzhang@ysu.edu.cn。
基金项目:国家自然科学基金资助项目(51271162);河北省高等学校科学技术研究项目重点项目(ZD2015048)
收稿日期:2015⁃03⁃12
文章编号:1007⁃791X(2015)03⁃0213⁃08
DOI:10.3969/j.issn.1007⁃791X.2015.03.004
文献标识码:A
中图分类号:TB303
猜你喜欢
北京大学学报(自然科学版)(2022年2期)2022-04-09
小哥白尼(趣味科学)(2020年6期)2020-05-22
电子制作(2019年10期)2019-06-17
电子制作(2019年24期)2019-02-23
NBA特刊(2018年21期)2018-11-24
北京汽车(2018年5期)2018-11-06
智富时代(2016年12期)2016-12-01
智富时代(2016年12期)2016-12-01
科技视界(2016年13期)2016-06-13
科技视界(2016年5期)2016-02-22
