
人血浆中甲氨蝶呤的高效液相色谱法测定及其代谢物的质谱定性分析
2015-12-29 01:24王漪璇,毋丹,曹军宁等
药学实践杂志 2015年2期
·论著·
人血浆中甲氨蝶呤的高效液相色谱法测定及其代谢物的质谱定性分析
王漪璇,毋丹,曹军宁,钱隽 (复旦大学附属肿瘤医院肿瘤内科,复旦大学上海医学院肿瘤学系,上海 200032)
[摘要]目的 建立固相萃取-高效液相色谱法测定人血浆中的甲氨蝶呤浓度,用于临床治疗的监测,并通过质谱对甲氨蝶呤的人体代谢物进行定性分析。方法 血浆样本经C18固相萃取小柱净化洗脱后直接进样,以Diamonsil C18分析柱(150 mm×4.6 mm, 5 m)分离,流动相为甲醇和0.5%乙酸溶液(含0.3%三乙胺),采用梯度洗脱;流速为1.0 ml/min;紫外检测波长为306 nm。通过质谱全扫描(QLMS)和多反应监测-信息关联采集-增强子离子扫描(MRM-IDA-EPI)获得甲氨蝶呤代谢物的质谱信息。结果 甲氨蝶呤在0.05~100 μmol/L范围内线性关系良好(r=0.999 9),提取回收率>95%,准确度在97%~105%之间,日内与日间精密度(RSD)均<5%。分析甲氨蝶呤代谢物的质谱信息,推断该代谢物为7-羟基甲氨蝶呤。结论 本方法灵敏度高、重现性好、操作简便,适用于甲氨蝶呤的血药浓度监测。
[关键词]甲氨蝶呤;高效液相色谱法;固相萃取;代谢物
[作者简介]王漪璇,中药师.E-mail:13564899963@139.com
[通讯作者]钱隽,副主任药师.研究方向:临床药理学.Tel:(021)64175590-81112;E-mail:junqian@fudan.edu.com
[中图分类号]R917.1[文献标志码]A
DOI[]10.3969/j.issn.1006-0111.2015.02.013
[收稿日期]2014-04-03[修回日期]2014-06-26
Determination of methotrexate in human plasma by HPLC and qualitative analysis of its metabolite by mass spectrometry
WANG Yixuan, WU Dan, CAO Junning, QIAN Jun(Department of Medical Oncology,Cancer Hospital Affilliated to Fudan University; Department of Oncology, Shanghai Medical College, Fudan University, Shanghai 200032, China)
Abstract[]ObjectiveTo develop an SPE-HPLC method for the determination of methotrexate (MTX) in human plasma to monitor the clinical drug use of MTX, and to identify the human metabolite of MTX by mass spectrometry.MethodsMTX was extracted from human plasma using C18 SPE column and analyzed directly after elution. The separation of MTX was performed on Diamonsil C18 column (150 mm×4.6 mm, 5 m) with a gradient mobile phase of methanol and 0.5% acetic acid solution (containing 0.3% triethylamine) at a flow rate of 1.0 ml/min. The detection wavelength was 306 nm. QLMS and MRM-IDA-EPI scan modes were used to obtain the mass spectrum of MTX metabolite.ResultsThe calibration curve of MTX in plasma was linear over the range of 0.05-100 μmol/L(r=0.999 9). The extraction recovery was above 95% and accuracy was between 97% and 105%. Both intra-and inter-day precision were less than 5%. 7-OH MTX was identified to be the metabolite through the analysis of the mass spectrum.ConclusionThe method is sensitive, reproducible, easy to operate and suits for monitoring the concentration of MTX in clinical practice.
[Key words] methotrexate; HPLC; solid phase extraction; metabolite
甲氨蝶呤(methotrexate, MTX)为抗叶酸类抗肿瘤药,临床上广泛用于白血病、淋巴瘤、骨肉瘤的化疗。但其毒性大且在人体内的代谢存在很大个体差异,当MTX给药后48 h血浆浓度在1.0 μmol/L、72 h血浆浓度在0.1 μmol/L以上时,会造成严重的甚至致命的毒性反应[1,2],必须及时给予亚叶酸钙(calcium folinate, CF)解救,因此大剂量使用MTX须及时监测患者的血药浓度。高效液相色谱法(HPLC)是治疗药物浓度监测的主要方法之一。已有很多文献[3-11]报道,应用HPLC法测定生物样品中MTX。本研究简化了原本烦琐的固相萃取过程,洗脱、浓缩一步完成且最低定量限达到0.05 μmol/L。同时收集患者血浆中MTX代谢物,通过质谱对其进行定性分析,确证了色谱中MTX代谢物为7-羟基甲氨蝶呤(7-OH MTX)。
1材料与方法
1.1仪器Agilent 1100 高效液相色谱仪、G1314A紫外检测器、HP Chem Station 工作站 V4.01(美国Agilent公司);API 3200 QTRAP三重四极杆串联质谱仪,配有电喷雾离子(ESI)源、操作软件Analyst 1.5(美国AB SCIEX 公司);Prominence UFLC液相色谱系统,包括LC-20AD梯度泵、SIL-20ACHT自动进样器、CTO-20A柱温箱和DGU-20A3脱气机(日本岛津公司);Bond Elut C18固相萃取小柱(100 mg/ml,美国Agilent公司)。
1.2药品与试剂MTX对照品(纯度99.6%,批号:100138-200603,中国药品生物制品检定所);对氨基苯乙酮(分析纯,批号:28295,上海晶纯试剂有限公司);甲醇(色谱纯,德国Merck公司);三乙胺和乙酸均为国产分析纯。
1.3HPLC色谱条件(MTX定量测定)色谱柱:Diamonsil C18柱(150 mm×4.6 mm, 5 m,Dikma);流动相A为0.5%乙酸溶液(含0.3%三乙胺),流动相B为甲醇,梯度洗脱见表1;流速:1.0 ml/min;柱温:25 ℃;紫外检测波长:306 nm;进样量:20 μl。
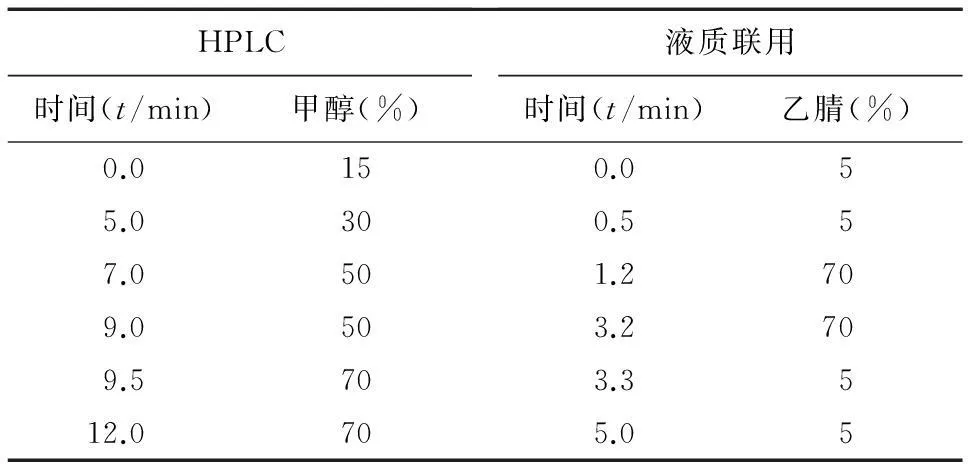
表1 梯度洗脱程序
1.4液质联用(代谢物定性分析)
1.4.1色谱条件色谱柱:Column Technology C18柱(50 mm×2.1 mm,3 m);流动相A为2 mmol/L乙酸铵,乙酸调pH值至4.0,流动相B为乙腈。梯度洗脱见表1;流速:0.3 ml/min;柱温:35 ℃。进样量:5 μl。
1.4.2质谱条件电喷雾离子源(ESI)正离子检测,采用全扫描(QLMS)和多反应监测-信息关联采集-增强子离子扫描(MRM-IDA-EPI),气帘气(CUR)压力为30 psi;雾化气(GS1)压力为35 psi;碰撞气(GS2)为40 psi,电喷雾电压(IS)为5 000 V。全扫描质量数范围:50~500;MRM选择监测离子对:m/z455→308、m/z471→324;IDA采集阈值:1 000 cps;EPI扫描速率:1 000 Da/s;扫描质量数范围:50~500。
1.5标准溶液的配制
1.5.1工作溶液精密称取11.36 mg的MTX对照品,加入甲醇溶解并定容,配制得1 000 μmol/L的MTX标准储备液。分别用50%甲醇稀释至0.50、1.00、2.50、10.0、25.0、100、250 μmol/L,作为MTX标准曲线工作液。吸取MTX 标准储备液分别用50%甲醇稀释至1.00、10.0、80.0、800 μmol/L,作为MTX质控点工作液。精密称取10.05 mg的对氨基苯乙酮对照品,加入甲醇溶解并定容,配制得5 g/L的内标储备液。将此溶液稀释50倍,获得浓度为100 mg/L的内标工作液。所有储备液和工作液均保存在4 ℃冰箱中备用。
1.5.2血浆标准曲线和质控样品向健康人空白血浆添加适量MTX系列工作溶液,配制成浓度为0.05、 0.10、 0.25、1.00、2.50、10.0、25.0、100 μmol/L的标准曲线样品。向人空白血浆添加MTX质控点工作液,得到浓度分别为0.10、1.00、8.00、 80.0 μmol/L的质控样品。
1.6血浆样品的处理
1.6.1液相进样样品在每毫升血浆样品中加入10 μl内标溶液,即获得含内标1 mg/L的载样血浆。取Bond Elut C18小柱,先以甲醇2 ml活化和超纯水2 ml平衡,继加入0.8 ml载样血浆,再以1 ml生理盐水淋洗,最后用60%甲醇溶液(含40 mmol/L 盐酸)0.4 ml洗脱。收集洗脱液涡旋5 s,HPLC进样20 μl。
1.6.2液质联用进样样品上述固相萃取洗脱液进样5 μl。收集患者样品HPLC进样10~11 min的洗脱液(即MTX代谢物收集液),混匀后进样5 μl分析,并与固相萃取洗脱液的质谱分析结果进行比较。
2结果
2.1特异性MTX和内标在本实验条件下保留时间分别为8.5和9.4 min。取6份不同来源的健康人空白血浆、空白血浆标准添加样品和实测样品,按“1.6.1”项下操作后进样分析,色谱图显示血浆中的内源性杂质均不干扰MTX和内标的测定,实测样品的MTX、内标和血浆样品中其他物质的色谱峰分离完全(图1)。
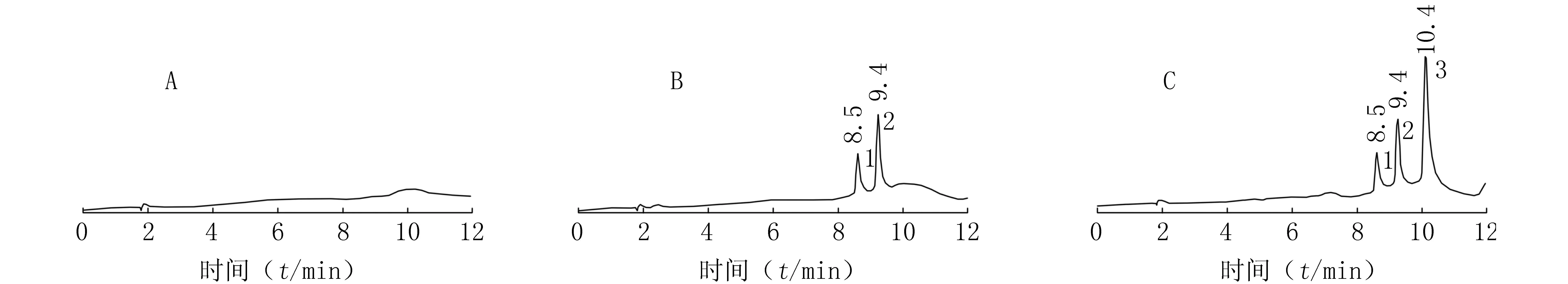
图1 人血浆中MTX的HPLC色谱图 A.空白血浆; B.空白血浆加MTX(1.00 μmol/L);C.患者用药48 h的实测血浆样品(1.26 μmol/L);1.MTX;2.内标;3.7-OH MTX
2.2线性范围与最低定量限浓度为0.05、0.10、0.25、1.00、2.50、10、25、100 μmol/L的血浆标准曲线样品,按“1.6.1”项下处理,经HPLC分析后,以MTX与内标的色谱峰面积之比(Y)对相应的浓度(X,μmol/L)进行线性加权(1/X2)回归,线性回归方程为:Y=0.278 5X-0.000 059,r=0.999 2。结果表明,MTX浓度在0.05 ~ 100 μmol/L范围内线性关系良好,最低定量限为0.05 μmol/L。
2.3回收率
2.3.1提取回收率取浓度为0.10、1.00、8.00、80.0 μmol/L的质控血浆样品平行制备5份,按“1.6.1”项下处理后进样测定。另直接用60%甲醇溶液(含40 mmol/L 盐酸)配制相应浓度的MTX溶液,进样20 μl,以质控样品与相应浓度MTX溶液的色谱峰面积之比,计算提取回收率,结果见表2。以同法测算内标回收率为93.2%。
2.3.2方法回收率4个浓度的质控样品平行制备5份,按“1.6.1”项下操作后进样测定,所得的MTX与内标的峰面积比以标准曲线方程计算,测得浓度与相应配制浓度的比值即为方法回收率,结果见表2。
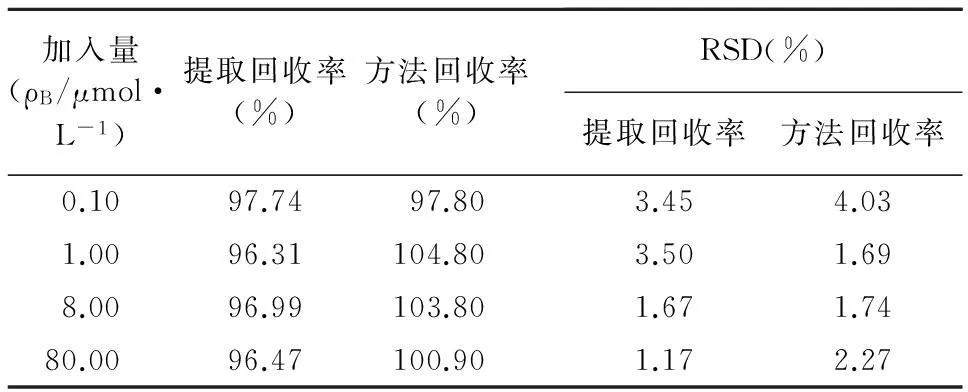
表2 MTX血浆样品的提取回收率和方法回收率( n=5)
2.4精密度4个浓度的质控样品平行制备5份,按“1.6.1”项下操作后测定,连续3 d同法处理进样,考察日内与日间精密度,结果表明本测定方法精密度良好(表3)。

表3 血浆样品中MTX浓度测定的精密度( n=5)
2.5稳定性
2.5.1样品的室温稳定性取浓度为0.10、1.00、8.00、80.0 μmol/L的质控血浆样品,在室温25 ℃下放置4 h后测定其浓度,结果与其理论浓度偏差<8.2%,RSD<7.2%,表明MTX于室温条件下4 h内稳定性良好。
2.5.2样品的冻融稳定性各浓度的质控血浆样品,反复冻融3次,每次冻融后分别测定,测得各冻融批次间样品的浓度变化<7.3%,RSD<7.8%,表明MTX血浆样品反复冻融3次稳定性良好。
2.5.3样品在自动进样器中的稳定性将各浓度的质控样品按“1.6.1”项下处理后立即进样,并在自动进样器中放置24 h后再次进样,比较前后两次进样测定浓度变化<4.3%,RSD<6.0%,表明MTX血浆样品经前处理后24 h内在自动进样器中稳定。
2.5.4样品存放期的稳定性将各浓度的质控血浆样品,在-40 ℃冰箱内存放3个月后测定其浓度,结果与其理论浓度偏差<8.7%,RSD<5.9%,证明MTX血浆样品在-40 ℃冰箱内存放3个月稳定性良好。
2.6稀释试验由于在临床治疗过程中,患者的MTX血药浓度值可能超过100 μmol/L,即超出本研究的定量上限,必须经过稀释后才能测定。用空白血浆将含1 000 μmol/L MTX的样品稀释20倍后测定,测得浓度乘20后与实际配制浓度比较,质控样品经过稀释后的准确度为101%,RSD<2.7%。
2.7临床应用本方法已应用于数百例使用大剂量MTX化疗患者的血药浓度监测。分析结果显示,MTX标准曲线和质控样品的重现性良好,患者血浆中内源性杂质、代谢物及合并用药均不干扰待测物峰。绝大多数患者在72 h的血药浓度已低于0.05 μmol/L,少数血药浓度不能下降至安全范围的患者在及时加大解救剂量和频率后血药浓度也降低至安全范围,均未发生不可逆的严重不良反应。
2.8代谢物鉴定固相萃取处理后的血浆样品经质谱全扫描可见m/z为455和471的离子,分别与MTX及其代谢物7-OH MTX的质荷比相吻合。根据文献报道[12,13],设置m/z455→308(MTX)及m/z471→324(7-OH MTX)的多反应监测,并对两组质荷比通道中的色谱峰进行IDA-EPI扫描,发现m/z471→324的通道中有色谱峰的保留时间为3.1 min,该物质的准分子离子为m/z471,主要的特征性碎片离子为m/z342、324、191,分别与MTX的准分子离子m/z455,特征性碎片离子m/z326、308、175各相差16(图2),证明两者在质谱中的碎裂方式相同(图3)。根据MTX的分子结构以及两者各碎片离子质荷比的差值均为16,推测前者在MTX的分子结构上发生了羟基取代,且取代位置为7号位,故推断其为MTX的代谢物7-OH MTX。MTX代谢物收集液通过液质联用进样后,在m/z471→324通道中色谱保留时间也为3.1 min,并在质谱中产生m/z324、191的碎片离子,证明在HPLC中保留时间为10.4 min的物质确为7-OH MTX。

图2 人血浆中MTX的HPLC色谱图MTX与7-OH MTX二级质谱图

图3 MTX与7-OH MTX的裂解模式
3讨论
3.1样品预处理HPLC色谱法测定生物样品中MTX,主要受样品预处理方法的限制,文献报道的蛋白沉淀[3-5]、液液萃取[6, 7]等方法均存在内源性杂质去除不完全而干扰待测物的测定、色谱柱污染严重、回收率低等问题[9,10]。本方法采用固相萃取法,基本去除了血浆样品中的基质干扰,方法特异性高,回收率达95%以上,重现性好,且省去了吹干复溶的步骤,操作简便迅速。
3.2固相萃取柱的选择本研究比较了Bond Elut C18、FOCUS、Bond Elut SAX和Bond Elut Plexa PCX等固相萃取柱的提取效率,结果显示Bond Elut C18柱回收率最高。经试验FOCUS柱洗脱液中MTX回收率略低于Bond Elut C18柱,极性杂质较C18柱多;离子交换柱(Bond Elut SAX、Bond Elut Plexa PCX)在保留、淋洗和洗脱的过程中均需调节pH值,且需预先去除血浆中竞争吸附的杂质方可载样,操作较为烦琐。
3.3内标的选择本实验考察了甲硝唑、茶碱、氨苯甲酸、安定、磺胺、对氨基苯乙酮、对氨基苯乙醚等化合物。结果表明,氨苯甲酸、安定、磺胺和对氨基苯乙醚的色谱保留时间与MTX差异较大,茶碱在紫外306 nm处吸收不佳,而甲硝唑在固相萃取过程中回收率较低。最后选择对氨基苯乙酮,其固相萃取的回收率较高,色谱保留时间与MTX相近但不干扰,且在紫外线检测波长306 nm处有较大吸收,适合作为MTX测定的内标化合物。
3.4样品浓缩由于临床上以用药72 h MTX浓度低于0.1 μmol/L为安全范围[1,2],所以MTX浓度的定量限应低于0.1 μmol/L。目前报道的固相萃取方法[8-11],均需吹干复溶进行浓缩方能够达到预期的定量限,此过程非常耗时。本研究通过减少固相萃取过程中洗脱液的用量,直接完成样品的浓缩。经试验在洗脱过程中使用不同体积的洗脱液(分别为0.2、 0.3、0.4、 0.5、0.7和1.0 ml),MTX的回收率分别为97.9%、99.3%、99.5%、99.1%、97.3%和99.0%,无明显差异,提示以少量洗脱液可同时完成洗脱和浓缩的过程,省去吹干复溶的操作,从而提高样品前处理的效率。
3.5质谱定性在定量分析过程中,对比空白加样血浆与患者血浆样品的液相色谱图,发现10.4 min时出现一色谱峰,见图1C,推测其或为MTX代谢物的色谱峰。由于实验室没有MTX代谢物的对照品,仅依靠HPLC无法确定此色谱峰的物质属性。API 3200 QTRAP是串联四级杆-线性离子阱质谱仪,其Q3可作为线性离子阱使用,在MRM扫描同时进行增强二级碎片的全谱定性。本研究采用MRM-IDA-EPI 组合扫描模式,
从患者用药后的血
浆样品中筛选出主要代谢物7-OH MTX,并获得液质联用的相关特征信息。经比较,色谱峰10.4 min处的物质在液质联用中的色谱与质谱行为与筛选所得7-OH MTX均相符,故确证其为MTX在人体内的代谢物7-OH MTX。
【参考文献】
[1]Green MR,Chowdhary S,Lombardi KM,etal.Clinical utility and pharmacology of high-dose MTX in the treatment of primary CNS lymphoma[J].Expert Rev Neurother,2006,6(5):635-652.
[2]Lennard L.Therapeutic drug monitoring of antimetabolic cytotoxic drugs[J].Br J Clin Pharmacol,1999,47:131-143.
[3]彭波.高效液相色谱法测定甲氨蝶呤的血药浓度[J].中国医院药学杂志,2011, 31 (11):951-952.
[4]张春燕,顾健.反相高效液相色谱法同时测定人血浆中亚叶酸、5-甲基四氢叶酸及甲氨蝶呤的浓度及临床应用[J].中国药学杂志,2010,45(7):543-547.
[5]许耘川,袁向亮,沈立松.HPLC法测定血清甲氨蝶呤浓度的方法建立及其与荧光偏振法的比较[J].检验医学,2010,25(9):701-704.
[6]谢军平,冯玲玲,匡霞.HPLC 测定大剂量甲氨蝶呤患者血药浓度[J].中国实验方剂学杂志,2010,16(11):69-72.
[7]霍晶,袁盛华,方芸,等.反相高效液相色谱法测定人血浆中甲氨蝶呤的浓度[J].药学与临床研究,2007,15(5):390-392.
[8]Turci R,Micoli G,Minoia C.Determination of methotrexate in environmental samples by solid phase extraction and high performance liquid chromatography: ultraviolet or tandem mass spectrometry detection?[J].Rapid Commun Mass Spectr,2000,14:685-691.
[9]周卿,潘雪君.固相萃取-高效液相色谱法测定人乳中甲氨蝶呤的含量[J].药物分析杂志,2011,31(12):2305-2307.
[10]吴雪梅, 丘宏强, 方令平, 等.固相萃取-高效液相色谱法测定甲氨蝶呤的血药浓度[J].中国医院药学杂志,2008,28(18):1617-1619.
[11]张华年,文玲莉,张少文, 等.固相萃取高效液相色谱法检测生物样本中甲氨喋呤[J].药物分析杂志,2000,20(6):401-404.
[12]曹一辰,孟祥乐,朱金辉,等.LC-MS/MS法测定人血清中甲氨蝶呤及其代谢产物7-羟基甲氨蝶呤的浓度[J].中国药师,2012,15(2):191-194.
[13]Guo PI, Wang XM, Liu LS,etal.Determination of methotrexate and its major metabolite 7-hydroxymethotrexate in mouse plasma and brain tissue by liquid chromatography-tandem mass spectrometry[J].J Pharm Biomed Anal,2007,43:1789-1795.
[本文编辑]李睿旻
猜你喜欢
现代临床医学(2022年4期)2022-09-29
中山大学学报(自然科学版)(中英文)(2022年4期)2022-08-05
健康体检与管理(2022年4期)2022-05-13
中国药学药品知识仓库(2022年7期)2022-05-10
现代临床医学(2021年2期)2021-03-29
分析化学(2016年7期)2016-12-08
分忧(2015年3期)2015-06-08
肉类研究(2015年1期)2015-04-08
大科技·百科新说(2014年5期)2014-06-10
中国民族民间医药·下半月(2011年10期)2011-12-27
