
超支化聚合物研究进展
2016-01-07 02:13卢苇萍,张春辉,钟荣
江西化工 2015年1期
超支化聚合物研究进展
卢苇萍张春辉钟荣
(南昌航空大学环境与化学工程学院,江西 南昌 330063)
摘要:超支化聚合物独特的三维立体球状结构使其具有低粘度、高流变性、良好的溶解性、大量可修饰的末端官能团和分子内部空穴结构等性质。本文综述了超支化聚合物结构、合成方法及应用,并提出了超支化聚合物的发展前景。
关键词:超支化聚合物结构研究进展
基金项目:*国家自然科学基金资助项目(项目编号:21364008)。
1引言
近年来,超支化聚合物由于其高度的支化结构,具备很多线性聚合物没有的性能,如低粘度、高流变性、良好的溶解性、大量可修饰的末端官能团和分子内部空穴结构[1]等独特的性质,这些特殊的性能让其在涂料方面、纳米材料方面、药物适缓剂方面和催化剂方面等多个领域显示出巨大的价值,已经逐渐成为人类生活中不可或缺的一部分。
早在1952年,Flory[2]发现了高度支化的聚合物可以通过 ABn(包括1个A官能团和2个或更多B官能团,n≥2)单体的自缩合反应而不产生凝胶来合成。非常遗憾的是,这种非结晶、无缠绕的聚合物当时并没有引起很多研究者的关注。直到1989年,杜邦公司Kim等[3]用 AB2型单体,3,5-二溴苯基硼酸或 3,5-二卤代苯基格氏试剂制得的超支化聚苯。1990年Kim[4]发表了关于超支化聚苯的论文并创造了超支化(hyperbranched)这一名词,超支化逐渐成为聚合物化学中的一个重要的分支及高分子研究界的热点。本文对超支化聚合物的结构、合成及应用进行了阐述。
2超支化聚合物的结构
2.1 结构
完美的树状大分子只有树枝状单元和末端的官能团,而超支化聚合物结构是从一个中心核出发,由支化单体 ABn(n≧2)逐级伸展,或者说超支化结构中有3种重复单元,即树枝状单元(Dendritic units)、线性单元(Linear units)和未反应的官能团B 决定的末端基团(Terminal units)(图 1)。
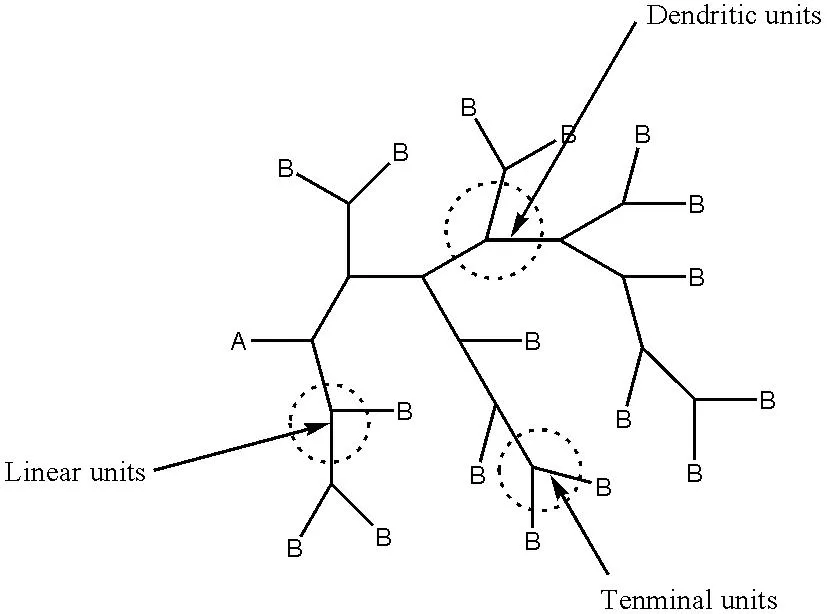
图1 超支化聚合物结构示意图
2.3 支化度
2.3.1定义
支化度 DB(degree of branching)作为表征超支化结构的一个重要因素,它标志着超支化聚合物和树枝状分子的接近程度,可以直接反映出聚合物结构的密度以及末端官能团的数目和位置。对于AB2单体缩聚形成的超支化聚合物,20 世纪 90 年代,Hawker和 Frechet[5]提出了计算支化度的公式。
DB=(D+T)/(D+L+T)
(1.1)
其中D、T、L 分别表示超支化聚合物分子中树枝状单元、末端基团以及线性结构单元所占的比例,完美树形超支化聚合物的DB=1。由于T=D+1,根据公式1.1可知,即使是完整的线性结构,DB>0。公式1.1可以准确地计算出具有高度支化聚合物的支化度,这种聚合物的树枝单元的数量和末端单元的数量差不多。而对于低摩尔质量的区域,就需要 Frey[6]提出的公式1.2,能够更好地计算支化度。
DB=1/(1+L/2D)
(1.2)
当L/D或者L/T能够很容易从核磁谱中计算出来,公式(1.2)就特别有用,但是经常是不能准确单元的数目。
2.3.2影响支化度的因素
Holter等人[7]分析AB2型单体无规缩聚过程中,研究了树形单元、末端单元和线型单元的形成过程,指出线型单元和末端单元、树形单元的活性、单体滴加的速度、滴加顺序、合成方法等都对支化度有影响。
3超支化聚合物的合成方法
3.1 自缩合乙烯基聚合(SCVP)
1995年,Frechet 及其合作者[8]提出用“自缩合乙烯基聚合”(SCVP)这个新方法来制备超支化聚合物。所用单体通式为AB,B活化后变成B*,作为引发中心。B*与另外一个单体的双键的加成完成链引发,形成有两个活性基团和一个双键的二聚体。每一个B*和新生成的A*都能与另一个分子的乙烯基以同样的方式反应所用单体是乙烯基基团带有一个活性基团,活性基团可以是阴离子、阳离子、自由基、和原子。该活性能够引发乙烯基增长,并且会迁移到下一个活性基团继续引发乙烯基聚合。反应历程如图2所示。

图2 自缩合乙烯基聚合合成机理
此后许多研究对SCVP法进行延伸。如氮氧基聚合(NMRP)[9,10]、原子转移自由基聚合(ATRP)[11,12]、可逆加成链转移聚合(RAFT)[13,14]加入到SCVP中。2012年,Mustafa Ciftci Muhammet U.Kahveci[15]等人提出了一个对SCVP改进的方法。此法采用的单体包含两种不同官能团,合成的超支化聚合物可以结合点击化学,合成很多功能性超支化聚合物。如丙烯酸炔丙基在自由基存在的情况下可以线性聚合,炔基在引发的情况下可以产生酯化点并且传递自由基。在聚合完后,在聚合物表面有很多没有反应完的炔基,可以用金属Cu(Ι)催化叠氮和其进行环加成反应[16,17],对超支化聚合物进行后改性。2014年,Uehara等人[18]用二乙烯基单体通过AFCT法(加成断裂链转移聚合)合成可以热固化的超支化聚合物,让自缩合乙烯基聚合更进了一步。
3.2 x单体缩聚法
ABx缩聚形成超支化聚合物是合成方法中最经典的一种。最早由 Flory[2]最早提出。这种方法凝胶的可能性比较小,用这种方法合成了聚苯[3,19]、聚醚[20]、聚酰胺[21]等超支化聚合物。开始时用AB2型单体,后来发展成了AB3[22]、AB4[23]甚至AB6[24]型单体。为了用ABx型单体成功的合成传统的超支化聚合物,ABx型单体的必须满足以下要求:官能团A和B在催化剂存在或经活化后可相互反应,并且没有副反应发生;官能团B的活性必须一致;官能团的活性不随反应的进行而变化;分子内不发生环化反应限制分子量。其合成机理如图三所示。由于大部分ABx单体不能商业化,所以限制了大规模的生产。第一例超支化聚苯由Kim等[3]利用缩聚法制得的,他们采用3,5-二溴苯基硼酸或3,5-二卤代苯基格氏试剂通过偶合反应合成端基为溴的超支化聚苯。Animesh[25]在2009发表了篇用AB2单体采用一步法合成可以功能化的超支化聚合物。AB2单体合成用三甲基苯酚溴甲基化后和炔丙醇反应得到。聚合后得到的超支化聚合物表面有很多的炔丙基,可以进行改性,用点击化学,和叠氮化合物点击反应。这样用AB2单体一步法合成的超支化聚合物可以改性使得超支化聚合物有很多的特殊性能,可以应用在很多行业上。
ABx缩聚合成超支化聚合物虽然简单,但是合成的产物结构难以控制,官能团无序分布,分子量分布很宽。而且ABx单体商业化的不多,大部分要自己合成,比较费时费力。为了更好的控制分子质量和结构,ABx单体优化成ABx+By型单体[26],合成的产物分散度也会大大降低。
3.3 开环聚合
开环超支化聚合(ROMBP)或者自冷凝开环超支化聚合(SCROP)和自缩合乙烯基聚合(SCVP)是很不同的。SCVP一般使用乙烯基单体,而开环聚合用的引发性单体是杂环化合物,如环氧乙烷类、ε-己内酯类、四氢呋喃类等等。目前利用开环聚合已可以用来合成超支化聚酰胺、聚醚、聚酯。Frey等人[27]利用缩水甘油进行阴离子聚合合成超支化聚合物,得到了分子量相对集中(PDI=1.1~1.4)的超支化聚合物。 2010年,Frey 等人[28]又对这种由缩水甘油开环聚合得到的超支化聚醚从合成到应用进行了系统的论述。开环聚合制备的超支化聚合物所用的单体和ABx、AB*类单体不同,所以用这个方法可制备一些新颖的超支化聚合物。
3.4 A 2+B y型单体聚合
由于ABx型单体大部分不能商业化,所以研究能够大规模生产的新类型的单体是热点。A2+By型单体就被提出来了。A2+By型单体缩聚与ABx型单体缩聚很不一样,很容易产生凝胶,可以用来制备交联聚合物材料。在1999年,Frechet[29]利用A2+B3的方法合成超支化脂肪族聚醚和聚酰胺。从那时起,大家开始意识到在凝胶点之前封端或者是特殊催化剂可以得到可溶性的超支化聚合物。2014年,孔杰等人[30]用A2+B3的方法合成超支化聚合物,并分析了描述超支化聚合物的一个新的参数-终端指数(Terminal Index)。
2012年,Wu等[31]用”A2+B4”的一锅法合成带有四苯乙烯的超支化聚合物,这种聚合物都表现出聚集诱导发光增强现象。
这种方法很难完全控制来获得分子量很大的聚合物,因为聚合物聚合物往往有部分凝胶的出现。所以关键研究在于如何控制不凝胶。产生凝胶有很多方面的因素,比如反应单体的浓度,光照强度,反应温度和反应时间等等。通过研究发现,避免凝胶的主要措施有:在凝胶点之前反映结束,在稀溶液中缓慢滴加单体。由于A2+By型单体聚合有这样的局限性,限制了其大规模工业化。现在有很多对A2+By型单体进行改进,如A2+A2′+B3体系。2007年,Wen等人[32]采用“A2+A2'+B3”方法合成了含有缺电子基团2,4,6-三(p-溴苯基)-1,3,5-三嗪的超支化聚芴。研究表明,投入单体的比率直接影响到超支化聚合物的溶解性,B3单体三嗪的比例越大,溶解性越差。
ABx型单体商业化原料较少,发展使用A2+By型单体,可是A2+By型单体容易凝胶,为了控制不凝胶,就要控制反应缓慢,溶液比较稀,或者是在凝胶点完成反应,这样增加了人工和成本,限制了其商业化。2000年。颜德岳[33]和Froehling研究组[34]分别提出偶合单体法制备超支化聚合物。偶合单体法是选择单体“A2+BB2”“A2+CBn”“AB+CDn”优先原位成ABx型单体,然后进一步反应成超支化聚合物。据Flory[2]报导ABx型单体缩聚成超支化聚合物凝胶的可能性不大,这样就可以避免凝胶出现。2009年,Li等人[35]使用多羟基胺和甲基丙烯酰氯原位生成ABn型的中间单体,然后迈克尔(Michael)加成聚合,使用N-异丙基丙烯酰胺封端后得到热敏型超支化聚(醚-胺)。
偶合单体法可以既得到能够商业化的ABx型单体,又能避免凝胶的出现,可以使超支化聚合物大规模的生产和使用。
3.5 其他的聚合方法
上述的一些方法是合成超支化聚合物的主要和常用的方法。除了那些方法外,还有一些不常用的方法。比如自组装聚合。自组装聚合经常用在药物医疗方面。Reinhoudt等[36]在1997年就开始用有机靶甲基氰化合物的可逆自组装合成超支化聚合物,这种聚合物加入乙腈后会降解。
4超支化的应用
超支化聚合物不同于线性聚合物,因其高度支化的超支化分子外围,存在很多末端基团,这些基团紧密堆积,使得中间成为一个密闭式的空腔结构,及大量可以改性的末端基团,使得超支化聚合具有低粘度、高流变性、良好的溶解性,这些让超支化聚合物在纳米材料方面、涂料方面、生物医疗方面等都有很大的应用。
4.1 纳米领域的应用
超支化聚合物具有比较特殊的结构,其内部或外部具有分散粒子的作用,不仅可以为小分子反应提供场所或模板,而且能使生成的纳米粒子稳定。近年来研究以超支化聚合物为模板制备纳米材料,通过控制超支化聚合物的结构和分子量的大小,从而合成不同尺寸的纳米材料。2006年,余木火[37]用乙酰基端基化的超支化聚合物纳米材料加入氨纶纺丝溶液中,快速搅拌几个小时后,氨纶纺丝液粘度降低。
2008年,Taniguchi[38]合成了能够接枝到多功能炭黑纳米粒子表面的超支化聚酯,并接枝成功,接枝率均达到60%。把超支化聚酯接枝到炭黑上,使炭黑粒子在四氢吠喃中能够形成稳定的胶体分散体系,改变了炭黑在溶剂中的分散性,也为制备端羟基多功能炭黑提供了一个简单易行的方法。
4.2 涂料方面的应用
超支化聚合物在UV涂料的应用中可以改善涂料的流动性,减少溶剂的用量,同时具有固化时间短、成膜性能优良等特点。近年来,环保需求越来越高,水性涂料尤其是UV水性涂料逐渐成为人们研究的热点,Asif[39]用丁二酸酐将十六端羟基的超支化脂肪族聚酯(BoltornTM-H2O)全部改性为羧端基,用甲基丙烯酸缩水甘油酯对产物部分改性,使超支化聚合物部分端基上包含了不饱和双键,得到了具有光固化特性的UV水性超支化聚酯,此超支化聚酯涂料能与商品涂料有很好的相容性。
2014年Cheng[40]用可溶性的丁二酸酐和以恶唑啉二醇单体缩聚成的具有羧酸类端基超支化聚合物,支化度达到60%。加入香茅醇在160℃反应产生具有白酒香味的树脂。动力学研究表明,超支化聚合物材料和这种已经商业化的醇酸树脂混合比只有醇酸树脂的涂料配方,粘性大大降低,而且干得特别快。
4.3 生物载体方面的应用
因超支化聚合物高度支化的超支化分子外围,存在很多末端基团,这些基团紧密堆积,使得中间称谓一个密闭式的空腔结构,可以保护很多客体分子的有效载荷,避免外界干扰。比如超支化聚合物作为药物缓释剂,可提高药物分子的溶解性,保护药物分子识别和穿透指定的细胞及降低到达靶细胞前降解。2009年,Chen等[41]通过共价键将阿司匹林包裹在以超支化聚酯为核外围接枝的新型药物释缓剂的内部。研究发现,在不同pH下,阿司匹林释放的时间不一样,可以通过这种对PH敏感的超支化聚酯来控制阿司匹林的释放速度。
超支化聚合物在生物医药方面研究有很大的进展。包括药物传送,基因转染,及蛋白质的检测,净化和传送等方面。2011年,Nakayma[42]制备了可以负载siRNA和DNA的超支化聚合物,发现分子量越大,支化数越多,PDI越小,传递性能越强,基因运送效率越高。
4.4 其他方面的应用
超支化聚合物的应用领域仍在不断扩大,如在智能材料[43]、分离膜[44]以及电致发光材料[32]等方面,其应用研究已成热点。
5结语
综上所述,超支化聚合物由于特殊的结构,所以具有良好的溶解性、低粘性和丰富的可以改性的末端结构等这些优异的性能,引起了很多研究者的关注。不断去研发新的方法合成超支化聚合物,新的可以商业化的单体,新的具有很多特点的超支化结构,这些都是以后的发展方向。控制超支化聚合物的分子量和支化度一直是研究者的难题。超支化聚合物的改性方面也是个热点,可以得到很多功能化的超支化聚合物等。超支化聚合作为一个新兴的研究领域,探索合成新方法、实现结构控制以及进一步的功能性研究方面都具有广阔的发展前景。
参考文献
[1]Ishizu K,Takahashi D,Takeda H.Polymer,2000.41(16):6081-6086.
[2]Flory P J.American Chemical Society,1952,74(11):2718-2723.
[3]Kim Y H.US,4857630,1987.
[4]Kim Y H,Webster O W.American Chemical Society,1990,112(11):4592-4593.
[5]Hawker C J,Lee R,Frechet J M J.American Chemical Society,1991.113(12):4583-4588.
[6]Frey H,Holter D.Acta Polym,1999,50(2-3):67-76.
[7]Frey H,Holter D.J Polym Mater 1997,77:226.
[8]Jean M J,Frechet,Masahiro H.et al.,Science,1995,269:1080-1083.
[9]Hawker C J,Bosman A W,Harth E.Chem Rev,2001,101(12):p.3661-3688.
[10]Peleshanko S,Gunawidjaja R,Petrash S et al.,Macromolecules,2006,39:4756-4766.
[11]Zou P,Yang L P,Pan C Y.J Polym Sci,Part A:Polym Chem,2008,46:7628-7636.
[12]Mei X,Liu A H,Cui J X,Wan X H,Zhou Q F.Macromolecules,2008,41:1264-1272.
[13]Tao L,Liu J Q,Tan B H,Thomas P Davis.Macromolecules,2009,42:4960-4962.
[14]Vogt A P,Sumerlin B S T.Macromolecules,2008,41:7368-7373.
[15]Ciftci M,Muhammet U Kahveci,Yagci Yet al.,Chem Commun,2012.48(82):10252-10254.
[16]Barner-Kowollik C,Du Prez F E,Espeel P et al.,Angew Chem,2011,50,60-62.
[17]Meldal,M.;Torn∅e,Chem C W.Chem Rev,2008,108,2952-3015.
[18]Sato E,Uehara I,Horibe H,Matsumoto A.Macromolecules,2014,47,937-943.
[19]Kim Y H,Webster O W.J Am Chem Soc,1990,112:4592-4593.
[20]Hong H Y,Mai Y Y,Zhou Y F et al.,J Polym Sci,Part A:Polym Chem,2008,46:668-681.
[21]Liou G S,Lin H Y,Yen H J.J Mater Chem,2009,19:7666-7673.
[22]Liu G T,Zhao M S.Polym Bull,2009,63:565-573.
[23]Wang L,He X H.J Poly Sci Part B:Polym Phys,2010,48:610-616.
[24]Miravet J F,Fréchet J M J.Macromolecules,1998,31:3461-3468.
[25]Saha A,Ramakrishnan S.Macromolecules,2009,42,4956-4959.
[26]Malmstroem E,Ohanson M,Hult A.Macromolecules,1995,28(5);1698-1703.
[27]Sunder A,Hanselmann R,Frey H,Mülhaupt R.Macromolecules,1999,32:4240-4246.
[28]Wilms D,Stiriba S E,Frey H.Accounts Chem Res,2010,43:129-141.
[29]Emrick T,Chang H T,Frechet J M J.Macromolecules,1999,32:6380-6382.
[30]Heng C,Jie K.Polymerization J Phys Chem,B2014,118,3441-3450.
[31]Wu W B,Ye S H,Huang L J et al.,J Mater Chem,2012.22(13):p.6374-6382.
[32]Wen G A,Xin Y,Zhu X R et al.,Polymer,2007.48(7):1824-1829.
[33]Yan D Y,Gao C.Macromol Symp,2000,151:581-589.
[35]Lin Y.Gao J W,Liu H W,Li Y S.Macromolecules,2009,42:3237-3246.
[36]Huck W T S,SnellinkRuel B H M,Lichtenbelt J W T et al.,Chem Commun.,1997(1):9-10.
[37]余木火,韩克清,李文江,等.中国专利:1766183.2006.
[38]Taniguchi Y,Ogawa M,Gang W et al.,Mater Chem Phys,2008,108(2-3):397.
[39]Asif A,Huang C Y,Shi W F.Polymers for Advanced Technologies,2003,14,609-615.
[40]Cheng G,Barnaby W,Greenland C et al.,Prog Org Coat,2014,77,1516-1522.
[41]Xia W,Jiang G H,Chen W X.J Appl Polym Sci,2008,109:2089-2094.
[42]Yasuhide,Nakayama,Acc Chem,2012,45,994-1004.
[43]Jia Z F,Chen H,Zhu X Y,Yan D Y.J.American Chemical Society,2006,128(25):8144-8145.
[44]We X Z,Zhu L P,Deng H Y et al.,J Membr Sci,2008.323(2):278-287.
The Prospect of Hyperbranched polymer
LU Wei-pingZHANG Chun-huiZHONG Rong
(DepartmentofMaterialsandChemistry,Control,SchoolofEnvironment&ChemicalEngineering,
NanchangHangkongUniversity,JiangxiNanchang330063China)
Abstract:Hyperbranched polymer has the low viscosity,high rheological property,good solubility and a large number of terminal groups which can be modified and the intramolecular hole structure,etc.Structure decides to property,property decides to application.The article shows the structure,the synthetic way,the application of hyperbranched polymer,and put forward its research prospect.
Key Words:Hyperbranched polymerStructureProspect
猜你喜欢
中学生数理化·高一版(2022年4期)2022-05-09
中学生数理化(高中版.高考数学)(2020年2期)2020-04-21
纤维复合材料(2018年2期)2018-12-07
铜仁学院学报(2018年6期)2018-07-05
衡阳师范学院学报(2016年3期)2016-07-10
材料科学与工程学报(2016年5期)2016-02-27
石油化工建设(2015年4期)2015-12-01
橡胶工业(2015年6期)2015-07-29
高中生学习·高二版(2015年3期)2015-05-21
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
