
CO2甲烷化催化剂与反应机理研究进展
2016-03-21 07:30赵云鹏田景芝郑钟植
天然气化工—C1化学与化工 2016年6期
赵云鹏,荆 涛,田景芝,郑钟植
(1.齐齐哈尔大学化学与化学工程学院,黑龙江 齐齐哈尔 161006;2.浦项工科大学化学工程系,韩国 浦项 790-784)
专论综述
CO2甲烷化催化剂与反应机理研究进展
赵云鹏1,荆 涛1,田景芝1,郑钟植2
(1.齐齐哈尔大学化学与化学工程学院,黑龙江 齐齐哈尔 161006;2.浦项工科大学化学工程系,韩国 浦项 790-784)
CO2甲烷化是有效利用二氧化碳资源的途径之一,是减少CO2的一种比较有效实际的方法,在环境保护方面显示出较大潜力。综述了Ni基催化剂、Ru基催化剂、Rh基催化剂等CO2甲烷化催化剂及其催化性能,以及催化反应机理的研究进展,展望了CO2甲烷化催化剂未来的发展方向。
二氧化碳;甲烷化;催化剂;反应机理
碳基能源的大量消耗使大气中CO2的浓度持续不断地增加,造成的温室效应得到了世界各国的广泛重视。近年来,对CO2的固定化技术方面的研究表现出很强的吸引力,其中以CO2为原料加氢合成甲烷、甲醇及二甲醚具有较好的发展前景;与形成其它碳氢化合物或醇类的反应相比较,CO2甲烷化的反应速度非常快,在反应热力学方面具有明显的优势[1,2]。因此,CO2甲烷化是减少CO2的一种比较有效实际的方法。反应所需的氢可通过利用多余的风电、光电通过电解等方法获得。此法,在德国、丹麦、日本等国已受到较大重视。
CO2甲烷化过程涉及的化学反应[3,4]:

CO2加氢合成甲烷的主反应(1)是放热的,而生成CO的逆水煤气变换副反应(2)是吸热的,从热力学分析来看,在通常条件下是有利于甲烷的生成,但随着温度的升高(>500℃),平衡向生成CO的方向移动。为了提高CO2甲烷化过程中CO2转化率和甲烷选择性,主要是通过对催化剂活性组分、助剂和载体的合理选择与组合来实现。目前,已报道的催化剂主要集中在负载型催化剂上,研究较多的金属活性组分有Ni、Ru、Rh基催化剂,其中对Ni基催化剂研究最多最普遍,还有其它的催化剂如Pd、Pt、Co等。常用的助剂和载体有Al2O3、SiO2、CeO2、ZrO2、La2O3、MgO、TiO2及分子筛等[1-7]。本文主要阐述了Ni、Ru、Rh基等催化剂上CO2甲烷化的催化性能,以及CO2甲烷化催化反应机理的研究进展。
1 Ni基催化剂
CO2甲烷化Ni基催化剂研究报道较多,催化剂的制备方法主要采用浸渍法,常用的载体有Al2O2、SiO2、CeO2、ZrO2、La2O3及分子筛等,Ni基催化剂上CO2的转化率和CH4的选择性均较好。
1.1 Al2O3为载体的催化剂
Riani等[8]研究了在Ni/Al2O3催化剂作用下,CO2加氢合成甲烷的性能。在523K的低温下,反应不能发生;随着反应温度的升高,CO2的转化率迅速增大,CH4的选择性增加比较缓慢;在773K时,CO2的转化率达到最大为71%,CH4的选择性为86%,CO的选择性为14%。
He等[9]研究了Ni-Al水滑石衍生催化剂(Ni-Al2O3-HT)上的CO2甲烷化反应,Ni-Al2O3-HT催化剂上Ni颗粒的大小分布窄,具有大量的强碱性位易于CO2的活化,促进了催化剂的反应活性,高度分散的Ni与强碱性载体的结合,使其对CO2甲烷化具有专一性和高效性。
Rahmani等[10]研究在介孔纳米γ-Al2O3载体上负载的 Ni催化剂中分别添加 CeO2,MnO2,ZrO2,La2O3助剂对CO2甲烷化反应的影响。TPR研究表明,在w(Ni)为20%的Ni/Al2O3催化剂上添加MnO2、CeO2助剂,使还原峰向低温方向移动,促进了Ni的还原。ZrO2、La2O3助剂的添加使还原峰向高温方向移动,抑制了Ni的还原。可见,MnO2、CeO2助剂改善了催化剂的还原性能。在350℃、空速9000mL/(g·h)、n(H2)/n(CO2)为3.5的反应条件下,w(Ce)为2%的Ce-Ni/Al2O3催化剂上CO2的转化率达到80.3%,CH4选择性为100%,并且在常压下连续反应600min,催化剂的活性和稳定性仍保持很好。
Liu等[11]通过浸渍法制备了w(Ni)为15%的Ni-CeO2/Al2O3催化剂,CeO2含量对催化剂的性能有较大的影响。在反应温度300℃、空速15000mL/(g·h)、n(H2)/n(CO2)为4的条件下,没有 CeO2存在的Ni/ Al2O3催化剂上CO2的转化率为45.0%;然而,在w(CeO2)为2%的Ni-CeO2/Al2O3催化剂上CO2的转化率迅速增大到71.0%;当w(CeO2)为4%时CO2的转化率几乎保持不变;继续增加到w(CeO2)为6%时,CO2的转化率略有降低,为68.0%。催化剂上大量的CeO2促进剂会逐渐降低催化剂的活性,这是由于其覆盖了Ni活性位而引起的。因此,w(CeO2)为2%~4%时,Ni-CeO2/Al2O3催化剂的性能最好。
Abello等[12]采用传统的金属硝酸盐共沉淀法制备较高Ni含量的Ni-Al混合氧化物Ni(Al)Ox催化剂(n(Ni)/n(Al)=5),获得较高的Ni含量和较大的催化剂比表面积。从理论上说,尽管较高的Ni含量(w(Ni) =70%)对于镍基催化剂的性能可能是不利的,但此研究的活化Ni-Al催化剂表现出较高的CO2转化率和CH4选择性,这主要是由于小的金属镍晶粒分散在 Ni-Al混合氧化物上。在 400℃、1MPa、n(H2)/ n(CO2)/n(N2)为4/1/1的反应条件下,CO2转化率达到92.4%,CH4选择性接近于100%。
1.2 SiO2为载体的催化剂
Aziz等[13]通过溶胶-凝胶法制备了介孔二氧化硅纳米粒子(MSN),然后以MSN为载体,采用浸渍法制备了Ni/MSN催化剂,并与Ni负载于其它载体如MCM-41、HY、SiO2及γ-Al2O3的催化剂进行了性能比较。结果表明,CO2甲烷化的反应活性从高到低的顺序为:Ni/MSN>Ni/MCM-41>Ni/HY>Ni/ SiO2>Ni/γ-Al2O3,这说明MSN作为甲烷化反应Ni基催化剂的载体具有较大的潜力,在甲烷化反应达到200h时,Ni/MSN催化剂仍保持很好的稳定性和催化活性,可显著抑制积炭的发生。XRD、N2吸附-脱附、吡咯吸附红外光谱研究表明[14],随着镍含量的增加,Ni/MSN催化剂的结晶度降低、表面积变小、碱性位减少。镍含量和碱性位浓度对催化剂保持高活性具有非常重要的影响。在623K、空速50000mL/(g·h)、n(H2)/n(CO2)为4的反应条件下,w(Ni)为1%和3%的Ni/MSN催化剂上 CO2的转化率分别为 11%和29%,在1mol Ni催化剂上甲烷的生成速率分别为3.45mol/s和9.00mol/s;w(Ni)为5%和10%的Ni/MSN催化剂上CO2的转化率显著增大,分别达到82%和85%,甲烷的生成速率迅速加快,在1mol Ni催化剂上甲烷的生成速率分别达到25.38mol/s和27.02mol/s。可见,随着镍含量增加,CO2的转化率和甲烷的生成速率都增大,但w(Ni)由5%增加至10%时,CO2的转化率和甲烷的生成速率增加较缓慢。催化剂上CO2甲烷化的反应活性从高到低的顺序为:10Ni/MSN≈5Ni/MSN>3Ni/MSN>1Ni/MSN。
1.3 分子筛为载体的催化剂
Westermann等[15]以USY分子筛为载体,采用浸渍法制备了w(Ni)分别为5%、10%和14%的Ni/USY催化剂,在250~450℃、空速43000 h-1、n(H2)/n(CO2)/ n(N2)为36/9/10且总流量250mL/min的反应条件下,w(Ni)从5%增加至14%,相应的CO2转化率从44.9%增大到72.6%,甲烷选择性从60%提高到95%。纯的USY分子筛载体并不具有CO2加氢反应活性。CO2转化率和甲烷选择性的提高主要是取决于催化剂外表面上负载的NiO数量,且在甲烷化反应之前,催化剂经过H2在470℃预处理,大部分的NiO先还原为金属Ni0。
Graca等[16]研究了以HNaUSY分子筛为载体负载Ni和Ce化合物的催化剂,Ni/USY催化剂上w(Ni)从2%增加至14%,CO2的转化率也随之增大,这是由于催化剂经过还原后,零价的Ni0物种数量较多的原因。在Ni/USY催化剂中添加Ce质量分数在3%~15%时,进一步改善了催化剂的活性和选择性,这是因为催化剂上的CeO2促进了CO2转化为CO,所以Ni-Ce/USY催化剂的性能是金属Ni活性位与CeO2促进剂之间协同催化作用的结果。
1.4 其它载体的催化剂
Tada等[17]采用浸渍法制备w(Ni)为10%的Ni/ CeO2和Ni/α-Al2O3催化剂,比表面积分别为46m2/g和7m2/g,H2-TPR表明在600℃不仅Ni被还原而且CeO2也得到了还原,CO2-TPD表明吸附在Ni/CeO2催化剂上的CO2数量更多,有利于CO2甲烷化反应。与Ni/α-Al2O3催化剂相比较,特别是在低温下Ni/CeO2催化剂上表现出较高的CO2转化率,CH4选择性接近100%。
Song等[18]采用浸渍法制备了w(Ni)为10%的高分散Ni/La2O3催化剂,在1.5MPa、空速3250 h-1、n(H2)/ n(CO2)为4的反应条件下,稳定反应2h后,反应温度从208℃升高至320℃,CO2转化率从4.5%提高到97.1%,甲烷的选择性都达到100%。在380℃、空速11000 h-1的条件下,CO2转化率为100%,甲烷的选择性为100%,甲烷的时空收率为1180g/(kg·h)。可见,Ni/La2O3催化剂对CO2甲烷化具有很好的催化作用。
Ocampo等[1]采用溶胶-凝胶法制备了不同镍含量的Ni-Ce0.72Zr0.28O2催化剂,5Ni-CZ(w(Ni)=5%)的比表面积最小为 70m2/g,NiO粒径最大为 26.3nm;10Ni-CZ(w(Ni)=10%)和15Ni-CZ(w(Ni)=15%)的比表面积较大分别为96m2/g和81m2/g,NiO粒径较小分别为11.5nm和13.8nm,说明10Ni-CZ和15Ni-CZ催化剂中NiO在Ce0.72Zr0.28O2载体上的分散性较好。实验表明,随着反应时间的增加,催化剂上CO2的转化率和CH4的选择性均降低,在反应150h时,10Ni-CZ催化剂上CO2的转化率为75.9%,CH4的选择性为99.1%,此催化剂的稳定性最好。
2 Ru基催化剂
Ru基催化剂对CO2甲烷化具有较好的催化性能,常用的助剂或载体有Al2O3、CeO2、ZrO2等。
Janke等[7]以Ru/γ-Al2O3为催化剂,在0.1MPa、空速4720h-1、n(H2)/n(CO2)为4的反应条件下,280℃基本达到了反应的热力学平衡状态,CO2的转化率和甲烷的选择性最好;随着反应空速增加,CO2的转化率降低并伴有CO的生成,甲烷的选择性下降;在高空速和高温时,CO的生成量也逐渐增多,这表明逆水煤气变换(RWGS)反应速率比CO加氢的反应速率快。在反应温度217℃、压力105Pa、空速4720h-1、n(H2)/n(CO2)为4/1的条件下,Ru/γ-Al2O3催化剂经过8次循环使用(总反应时间72h),没有CO生成,其仍保持较好的催化活性和甲烷的选择性,无失活的现象发生。
Sharma等[19]研究了氧化铈掺杂Ru的催化剂上进行CO2甲烷化的反应。在气体组成为Ar 5mL/min、 CO22mL/min、H28mL/min,450℃的条件下,Ce0.96Ru0.04O2和 Ce0.95Ru0.05O2的催化性能最好,CO2转化率为55%,甲烷选择性为99%。随着Ce1-xRuxO2催化剂中Ru掺杂量的增加,CO2转化率和甲烷选择性升高,当x>0.03时,CO2转化率增加缓慢,甲烷选择性没有变化。
Tada等[20]研究了Ru/CeO2/Al2O3催化剂上CeO2质量分数对CO2甲烷化反应的活性和CH4选择性的影响。Ru/Al2O3催化剂中添加CeO2,CO2的反应速率增大。在250℃时,CO2的反应速率由大到小的顺序为:Ru/30%CeO2/Al2O3(6.2mL/(min·g))>Ru/60% CeO2/Al2O3(4.9mL/(min·g))>Ru/CeO2(4.6mL/(min·g))> Ru/Al2O3(1.2mL/(min·g))。可见,Ru/CeO2/Al2O3催化剂上具有较好的CO2甲烷化反应活性,而且甲烷的选择性高于Ru/CeO2催化剂上甲烷的选择性。研究表明Ru/CeO2/Al2O3催化剂上的CeO2高度分散在Al2O3上,部分CeO2被还原,促进了CO2的甲烷化。
Li等[21]采用均相沉淀法在不同的焙烧温度下制备了Ce0.8Zr0.2O2载体,然后通过浸渍法制备了Ru为活性组分的 Ru/Ce0.8Zr0.2O2催化剂。研究表明在500℃下进行焙烧的Ce0.8Zr0.2O2载体上形成了Ce-Zr固溶体,具有适宜的表面积和孔结构,并且与活性组分Ru之间具有较弱的相互作用,催化剂的活性得到显著提高。以500℃下焙烧的Ce0.8Zr0.2O2为载体,制备的Ru/Ce0.8Zr0.2O2催化剂在400℃下焙烧,并连续经过H2N·NH2·H2O和H2还原,其具有较高的CO2甲烷化反应催化活性。在290℃、0.1MPa、空速10000h-1、n(H2)/n(CO2)为3.5的反应条件下,原料气H2的转化率达到93.57%。
3 Rh基催化剂
Rh基催化剂是CO2甲烷化最有效的催化剂之一,目前采用的催化剂载体主要是Al2O3。同时,报道了添加Ba和K对Rh/Al2O3催化剂性能的影响。
Beuls等[22]研究了Rh/γ-Al2O3催化剂上CO2甲烷化的过程,CO2加氢生成甲烷的选择性为100%,生成的甲烷数量不仅取决于温度、压力,还与原料气中加入CO或O2气体促进剂有关,实验表明在低温低压下可以生成甲烷产物,原料气中CO的加入抑制了CO2甲烷化,原料气中加入少量的O2能够促进CO2的甲烷化,而加入过多的O2不利于CO2的甲烷化。
Karelovic等[23]研究了不同Rh含量的Rh/γ-Al2O3催化剂上低温条件下CO2甲烷化的反应性能。与载体γ-Al2O3相比较,负载Rh的Rh/γ-Al2O3催化剂的比表面积仅有微小的变化,而孔容增大,其中w(Rh)为2%的Rh/γ-Al2O3催化剂孔容最大为0.38cm3/g。Rh的分散性取决于Rh/γ-Al2O3催化剂上Rh的含量,w(Rh)为1%的Rh/γ-Al2O3催化剂上Rh的分散性最好,而催化剂上w(Rh)为大于3%时,Rh的分散性基本稳定不变化。随着催化剂上w(Rh)从1%增加至5%,Rh的平均颗粒大小从3.6nm增大到15nm左右,其中w(Rh)为3%的Rh/γ-Al2O3催化剂上Rh的平均颗粒大小达到15.4nm。在大气压力、反应温度135~200℃条件下,Rh/γ-Al2O3催化剂上CH4的选择性均为100%。
Buchel等[3]研究了在Rh/Al2O3催化剂上添加Ba和K对CO2加氢的影响,Ba主要以BaCO3的形式存在,而K以KHCO3和KOH的形式存在。在Rh/ Al2O3催化剂、含Ba的Rh/Al2O3催化剂上,低于500℃时,CH4的选择性较高,并且在400℃时,CH4的产率最大为60%;在400℃以上时,根据热力学平衡关系可知,产物以逆水煤气变换反应生成的CO和H2O为主。在含K的Rh/Al2O3催化剂上,300~800℃的全部反应温度范围内都没有甲烷生成,CO2全部都转化为CO。
4 其它催化剂
除了上述Ni、Ru、Rh基催化剂,其它活性组分的催化剂有Pt、Pd、Co等,常用的助剂和载体有TiO2、MgO、SiO2、MCM等。这些催化剂对CO2甲烷化也具有较好的催化性能。
Yu等[24]通过碱水热法合成TiO2纳米管作为载体,并与Pt的络合物进行光化学沉积制备了Pt/ TiO2纳米管催化剂(Pt/Tnt)。Pt/Tnt催化剂具有较大的比表面积,为187m2/g。由TPR和XPS表征可知,混合价态的1~3nm Pt纳米颗粒均匀地分散于多壁的TiO2纳米管上。CO2-TPD结果表明大量的CO2吸附在Pt/Tnt催化剂上,Pt/Tnt催化剂具有较好的吸附CO2的能力,这主要是由于大比表面的纳米管结构与混合价态的Pt纳米颗粒之间协同作用的结果。原位红外光谱证实在100℃的低温条件下,Pt/Tnt催化剂具有较高的CO2加氢合成甲烷的催化活性,因此Pt/Tnt催化剂对CO2回收利用及加氢合成甲烷过程是非常有发展潜力的。
Park等[25]采用反相微乳液法制备了高分散的Pd-Mg/SiO2催化剂,经过焙烧后形成5~10nm的Pd颗粒分散在无定形的Mg和Si氧化物上。在反应温度450℃、原料气流量10.2cm3/min、n(H2)/n(CO2)为4的条件下,Pd-Mg/SiO2催化剂上CH4的选择性为95%,CO2的转化率为59%。不含Mg的Pd/SiO2催化剂仅对CO2还原为CO有很好的活性,Mg/SiO2催化剂对甲烷化反应的活性很差。研究表明Pd和Mg/ Si的氧化物对CO2甲烷化具有较好的协同作用。
Janlamool等[26]研究了CoTiMCM催化剂上CO2加氢合成甲烷的反应,焙烧后的催化剂上Ti是以锐钛矿型TiO2的形式存在的。TiO2对催化剂的性能具有非常重要的作用,主要表现在:活性组分Co的氧化物与TiO2载体之间的相互作用促进了Co的还原,阻止了硅酸盐化合物的形成,抑制逆水煤气变换反应,降低CO的选择性。与CoMCM催化剂相比较,CoTiMCM催化剂表现出较好的催化活性。在220℃、101.325kPa、n(H2)/n(CO2)/n(Ar)为20/2/8、原料气流量18L/(g·h)的反应条件下,反应6h后,CO2的转化率为34%,甲烷的选择性为94.9%,CO的选择性为5.1%。
5 CO2甲烷化反应机理
目前,对CO2甲烷化提出的反应机理主要有两种类型:一个是在甲烷化之前CO2转化为CO,由CO甲烷化生成甲烷;另一个是CO2直接加氢合成甲烷,并没有CO中间体的形成。大多数的研究主要倾向于第一个反应机理,然而,对于中间体的性质以及甲烷形成的过程仍存在不同的观点。
Aziz等[27]研究了在介孔二氧化硅纳米颗粒(MSN)上负载金属组分 (Ni、Rh、Ru、Fe、Ir、Cu)催化剂上CO2甲烷化反应机理,如图1所示。通过原位红外光谱分析表明了金属、MSN以及金属/MSN在CO2甲烷化反应中的作用。首先,CO2和H2在金属位上吸附与解离,形成CO、O和H原子,随后向MSN表面迁移,解离生成的CO与MSN表面上的氧作用形成桥式和线性羰基,而H原子的存在促进了双齿甲酸盐的形成。同时,O原子溢流到MSN表面,并稳定在金属位附近的氧空位上,在MSN表面上吸附的O与H原子反应生成OH基,OH基再与另一个H原子反应生成H2O。吸附的含碳物种进一步加氢生成CH4和H2O。

图1 金属/MSN催化剂上CO2甲烷化反应机理Fig.1 Reaction mechanism for CO2methanation over metal/MSN catalyst
Pan等[28]通过原位红外光谱对CO2分别在Ni/ Ce0.5Zr0.5O2、Ni/γ-Al2O3催化剂上的吸附和甲烷化反应过程进行了研究。与Ni/γ-Al2O3催化剂相比,Ni/ Ce0.5Zr0.5O2催化剂表现出较好的CO2甲烷化活性,在这两种催化剂上的反应路径是相似的,而仅在于碱性位作用的不同。Ni/Ce0.5Zr0.5O2催化剂具有弱和中等强度的碱性位,而Ni/γ-Al2O3催化剂具有弱和强碱性位。CO2吸附在Ni/Ce0.5Zr0.5O2催化剂的中等强度碱性位上形成单齿配位碳酸盐,而吸附在Ni/γ-Al2O3催化剂强碱性位上的CO2没有参与甲烷化反应。在Ni/Ce0.5Zr0.5O2催化剂上,中等强度碱性位上的单齿配位碳酸盐进一步形成单齿配体甲酸盐,此单齿配体甲酸盐加氢反应速率比由碳酸氢盐形成的双齿配体甲酸盐加氢反应速率快。中等强度的碱性位促进了单齿配体甲酸盐的形成,因此催化剂活性更好。
Karelovic等[29]通过原位漫反射红外光谱实验研究表明,在50℃低温时Rh/γ-Al2O3催化剂上CO2容易发生吸附与解离,形成线性的Rh-CO物种,桥式的Rh2(CO)和Rh2(CO)3物种,随着催化剂上Rh颗粒的增大,Rh2(CO)和Rh2(CO)3物种的数量也增多。然而,在所形成物种中以线性的Rh-CO物种为主,Rh-CO物种与H结合形成Rh的羰基化合物。吸附的CO是合成甲烷非常重要的中间体,甲酸盐对甲烷的生成没有作用,而且发现CO解离的活化能与合成甲烷反应的活化能非常接近,这表明反应过程中CO键的解离非常重要。
Park等[25]提出了 Pd-Mg/SiO2催化剂上CO2甲烷化反应机理,如图2所示。吸附在Pd上的H原子溢流到由CO2和MgO形成的碳酸盐表面上,然后依次逐步加氢形成甲烷,并最终从催化剂表面上脱附得到甲烷产物。可见,经过一个加氢循环后的MgO再次与CO2结合形成碳酸盐,继续与从吸附在Pd上的溢流H原子加氢反应,实现又一个新的加氢循环过程。
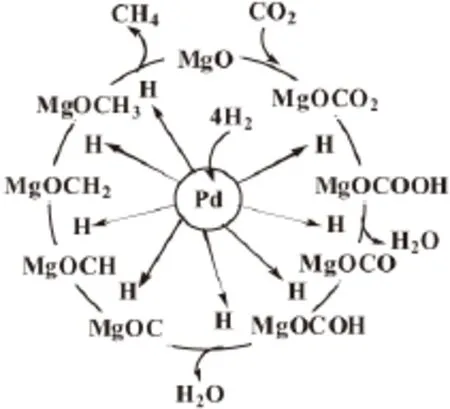
图2 Pd-Mg/SiO2催化剂上CO2甲烷化反应机理Fig.2 Reaction mechanism for CO2methanation over Pd-Mg/SiO2catalyst
6 结语
近年来,CO2甲烷化反应得到了比较广泛的关注和研究,主要体现在催化剂的种类、催化剂的制备方法、催化反应机理等方面,特别是在Ni、Ru、Rh基等催化剂性能以及浸渍法制备催化剂的方法上取得了一系列研究成果,不同种类的催化剂上甲烷的选择性普遍较高。在催化反应机理方面,大多数的研究认为有CO中间体生成,在不同组分的催化剂上形成的反应中间物种主要有碳酸盐、甲酸盐或羰基化合物。研究还发现,介孔二氧化硅纳米颗粒(MSN)负载金属组分(如Ni、Ru、Rh等)催化剂上的MSN载体对CO2甲烷化具有重要作用,MSN载体上形成的羰基物种是生成甲烷的前驱体,这为CO2的催化研究提供了新的观点,也便于更好地理解CO2甲烷化的反应机理。综上所述,提高催化剂上CO2的转化率,改善催化剂的制备方法,进一步明确认识CO2甲烷化反应形成的中间体以及由中间体最后生成甲烷的过程是未来发展的主要方向。从环境保护和能源化学两个方面上考虑,CO2甲烷化反应过程将具有非常广阔的发展与应用前景。
[1]Ocampo F,Louis B,Roger A.Methanation of carbon dioxide over nickel-based Ce0.72Zr0.28O2mixed oxidecatalysts prepared by sol-gel method[J].Appl Catal A, 2009,369:90-96.
[2]Akamaru S,Shimazaki T,Kubo M,et al.Density functional theory analysis of methanation reaction of CO2on Ru nanoparticle supported on TiO2(1 0 1)[J].Appl Catal A,2014,470:405-411.
[3]Buchel R,Baiker A,Pratsinis S E.Effect of Ba and K addition and controlled spatial deposition of Rh in Rh/ Al2O3catalysts for CO2hydrogenation[J].Appl Catal A, 2014,477:93-101.
[4]JimenezV,SanchezP,Panagiotopoulou P,etal. Methanation of CO,CO2and selective methanation of CO,in mixtures of CO and CO2,over ruthenium carbon nanofibers catalysts[J].Appl Catal A,2010,390:35-44.
[5]Cai M D,Wen J,Chu W,et al.Methanation of carbon dioxide on Ni/ZrO2-Al2O3catalysts:Effects ofZrO2promoter and preparation method of novel ZrO2-Al2O3carrier[J].J Nat Gas Chem,2011,20:318-324.
[6]Guo M,Lu G X.The effect of impregnation strategy on structural characters and CO2methanation properties over MgO modified Ni/SiO2catalysts[J].Catal Commun,2014, 54:55-60.
[7]Janke C,Duyar M S,Hoskins M,et al.Catalytic and adsorption studies for the hydrogenation of CO2to methane[J].Appl Catal B,2014,152-153:184-191.
[8]Riani P,Garbarino G,Lucchini M A,et al.Unsupported versus alumina-supported Ni nanoparticles as catalysts for steam/ethanol conversion and CO2methanation[J].J Mol Catal A,2014,383-384:10-16.
[9]He Li,Lin Q Q,Liu Y,et al.Unique catalysis of Ni-Al hydrotalcite derived catalyst in CO2methanation: Cooperative effect between Ni nanoparticles and a basic support[J].J Energ Chem,2014,23:587-592.
[10]Rahmani S,Rezaei M,Meshkani F.Preparation of promoted nickel catalysts supported on mesoporous nanocrystalline gamma alumina forcarbon dioxide methanation Reaction[J].J Ind and Eng Chem,2014, 20:4176-4182.
[11]Liu H Z Zou X J,Wang X G,et al.Effect of CeO2addition on Ni/Al2O3catalysts for methanation of carbon dioxide with hydrogen[J].J Nat Gas Chem,2012,21: 703-707.
[12]Abello S,Berrueco C,Montane D.High-loaded nickelalumina catalystfordirectCO2hydrogenation into synthetic natural gas(SNG)[J].Fuel,2013,113:598-609.
[13]Aziz M A A,Jalil A A,Triwahyono S,et al.Highly active Ni-promoted mesostructured silica nanoparticles for CO2methanation[J].Appl Catal B,2014,147:359-368.
[14]Aziz M A A,Jalil A A,Triwahyono S,et al.CO2methanation overNi-promoted mesostructured silica nanoparticles:Influence of Ni loading and water vapor on activity and response surface methodology studies[J]. Chem Eng J,2015,260:757-764.
[15]Westermann A,Azambre B,Bacariza M C,et al.Insight into CO2methanation mechanism over NiUSY zeolites: An operando IR study[J].Appl Catal B,2015,174-175: 120-125.
[16]Graca I,Gonzaez L V,Bacariza M C,et al.CO2hydrogenation into CH4on NiHNaUSY zeolites[J].Appl Catal B,2014,147:101-110.
[17]Tada S,Shimizu T,Kameyama H,et al.Ni/CeO2catalysts with high CO2methanation activityand high CH4selectivity at low temperatures[J].Int J Hydrogen Energ, 2012,37:5527-5531.
[18]Song H L,Yang J,Zhao J,et al.Methanation of Carbon Dioxide over a Highly Dispersed Ni/La2O3Catalyst[J]. Chin J Catal,2010,31:21-23.
[19]Sharma S,Hu Z P,Zhang P,et al.CO2methanation on Ru-doped ceria[J].J Catal,2011,278:297-309.
[20]Tada S,Ochieng O J,Kikuchi R,et al.Promotion of CO2methanation activity and CH4selectivity at low temperatures over Ru/CeO2/Al2O3Catalysts[J].Int J Hydrogen Energ,2014,39:10090-10100.
[21]Li T,Wang S,Gao D N,et al.Effect of support calcination temperature on the catalytic properties of Ru/Ce0.8Zr0.2O2for methanation of carbon Dioxide[J].J Fuel Chem Technol,2014,42:1440-1446.
[22]Beuls A,Swalus C,Jacquemin M,et al.Methanation of CO2:Further insight into the mechanism over Rh/γ-Al2O3catalyst[J].Appl Catal B,2012,113-114:2-10.
[23]Karelovic A,Ruiz P.CO2hydrogenation at low temperature over Rh/γ-Al2O3catalysts:Effect of the metal particle size on catalytic performances and reaction mechanism[J]. Appl Catal B,2012,113-114:237-249.
[24]Yu K P,Yu W Y,Kuo M C,et al.Pt/titania-nanotube:A potential catalyst for CO2adsorption and hydrogenation [J].Appl Catal B,2008,84:112-118.
[25]Park J N,McFarland E W.A highly dispersed Pd-Mg/ SiO2catalyst active for methanation of CO2[J].J Catal, 2009,266:92-97.
[26]JanlamoolJ,Praserthdam P,JongsomjitB.Ti-Si composite oxide-supported cobaltcatalystsforCO2hydrogenation[J].J Nat Gas Chem,2011,20:558-564.
[27]Aziz M A A,Jalil A A,Triwahyono S,et al.Methanation of carbon dioxide on metal-promoted mesostructured silica nanoparticles[J].Appl Catal A,2014,486:115-122.
[28]Pan Q S,Peng J X,Sun T J,et al.Insight into the reaction route of CO2methanation:Promotion effect of medium basic sites[J].Catal Commun,2014,45:74-78.
[29]Karelovic A,Ruiz P.CO2hydrogenation at low temperature over Rh/γ-Al2O3catalysts:Effect of the metal particle size on catalytic performances and reaction mechanism[J]. Appl Catal B,2012,113-114:237-249.
Progress in catalysts and reaction mechanisms for CO2methanation
ZHAO Yun-peng1,JING Tao1,TIAN Jing-zhi1,CHUNG Jong-shik2
(1.College of Chemistry and Chemical Engineering,Qiqihar University,Qiqihar 161006,China; 2.Department of Chemical Engineering,Pohang University of Science and Technology,Pohang 790-784,Republic of Korea)
CO2methanation is one of approaches to effectively utilize CO2resource and reduce CO2emission,showing great potential in environmental protection.The research progresses in the catalysts which could be used for CO2methanation,including Ni-based,Ru-based,Rh-based catalysts,etc.,and their catalytic performances as well as the reaction mechanisms are reviewed. Future development directions on CO2methanation catalysts are pointed out.
carbon dioxide;methanation;catalyst;reaction mechanism
O643.3;TQ426;TQ221.11
:A
:1001-9219(2016)06-98-07
2016-04-20;
:黑龙江省教育厅科学技术研究项目(12511596);
:赵云鹏(1973-),男,博士,副教授,电邮zhyypp@163.com。
猜你喜欢
机械工业标准化与质量(2022年6期)2022-08-12
航空维修与工程(2022年11期)2022-02-06
建材发展导向(2021年14期)2021-08-23
缔客世界(2020年10期)2020-04-10
中国煤层气(2019年2期)2019-08-27
中国调味品(2017年2期)2017-03-20
中国新技术新产品(2017年3期)2017-03-07
中学化学(2015年2期)2015-06-05
理科考试研究·高中(2014年8期)2014-10-17
火炸药学报(2014年1期)2014-03-20
