
金属掺杂类石墨相氮化碳的理论研究
2016-05-12 00:55王海燕
化学研究 2016年2期
关键词:能隙
李 鹏,王海燕,朱 纯
(贵州大学 化学化工学院, 贵州 贵阳 550025)
金属掺杂类石墨相氮化碳的理论研究
李鹏,王海燕*,朱纯*
(贵州大学 化学化工学院, 贵州 贵阳 550025)
摘要:采用量子化学从头算密度泛函理论(DFT)计算方法,对不同金属元素掺杂的M/g-C3N4(M=Mn, Cu, Au)构型进行优化,分析比较这些结构的能隙大小、金属原子的结合能以及前线轨道. 结果表明:M/g-C3N4(M=Mn, Cu, Au)的稳定性顺序为Mn/g-C3N4>Cu/g-C3N4>Au/g-C3N4;掺杂几种金属原子后M/g-C3N4(M=Mn, Cu, Au)的能隙明显减小,极大的增强了g-C3N4在可见光范围吸收能力,提高了g-C3N4的光催化效率. 本研究对进一步理解结构修饰对g-C3N4光催化性能的影响提供了一定的理论支持.
关键词:密度泛函理论;光催化材料; g-C3N4;金属掺杂;能隙
进入21世纪以来,传统能源短缺和环境污染已成为制约经济发展的主要问题,研究开发清洁、高效、可持续的新能源成为人类迫切需要解决的难题. 太阳能作为新能源的一种,因其具有洁净、安全、廉价、无污染等优点而越来越受到人们的重视. 1972年,FUJISHIMA等[1]在Nature 杂志上发表了关于半导体TiO2单晶电极在可见光下能够催化电解水的研究, 这一研究开启了多相光催化利用太阳能的新途径,标志着光催化研究的开始. 当前,市场上最主要的光伏材料依然是晶体硅材料,其总光电转换效率可达到 20%~24%[2-3]. 尽管单晶硅电池具有电池转换效率高、稳定性好的特点,但由于单晶硅生产成本居高不下,对环境也有很大的污染和破坏,因此探索开发新型高效的太阳能光催化材料已成为目前光催化技术领域的研究热点之一[4-6].
1989 年, LIU 和 COHEN[7]首次从理论上预测了具有与金刚石相当硬度的β相氮化碳(β-C3N4)材料. 1996年,TETER和HEMLEY[8]运用第一性原理对C3N4进行了进一步的计算,推测出C3N4可能的5种晶相, 即α相、β相、立方相、准立方相和类石墨相(g-C3N4). 计算结果表明,常温常压下,g-C3N4是最稳定的相,如图1[9]所示,即3-s-三嗪(C6N7),环与环之间通过中间的N原子形成无限扩展的二维平面.

图1 构成g-C3N4的基本结构单元3-s-三嗪Fig.1 Sym-triazine unit of g-C3N4
2009年,WANG等[10]研究发现在可见光作用下,g-C3N4可以光催化分解水制氢,这一研究结果进一步将g-C3N4的应用扩展至多相光催化的研究领域. g-C3N4因其具有不溶于水、化学稳定性好、廉价易得、节能环保等优点[11],使其在环境污染治理[12]、清洁能源再生[13]等领域具有很广阔的应用前景,因此也越来越受到人们的关注[14-15]. g-C3N4的禁带宽度较大,理论值约为2.70 eV,导致它仅能吸收超过450 nm的紫外-可见光,而紫外光在太阳光中只占极小的比例 (约为4%)[16]. 另外,由于g-C3N4自身比表面积较小和光生载流子易复合等缺陷,极大地降低了g-C3N4的光催化效率. 目前,通过向g-C3N4中掺杂其他元素、化合物对其进行结构的改性,以降低光生载流子复合几率,提高g-C3N4的光催化性能已取得了一定的研究进展[17-18]. 硫掺杂的g-C3N4扩大了g-C3N4对可见光的响应范围[19-20],光催化水解效率提高了7~8倍. 硼掺杂的g-C3N4材料[21]热稳定性增强,可见光响应范围增大光催化活性得到提高. 阮林伟等[22]使用第一性原理计算发现g-C3N4掺杂B、 P、 S三种非金属元素后体系的禁带宽度降低,拓展了其光吸收阈值,并在红外光区有很强的吸收,明显提高了光催化效率. 金瑞瑞等[23]成功制备了铁掺杂石墨相氮化碳(Fe/g-C3N4),铁元素的掺杂改变了g-C3N4的电子结构,抑制了光生电子-空穴对的复合,表现出优异的光催化性能. YE等[24]成功合成了Fe2O3/g-C3N4复合光催化材料,与纯的g-C3N4相比,Fe2O3/g-C3N4复合材料禁带宽度减小,表现出很强的光吸收性能. 另外,g-C3N4还可以和其他功能性材料结合,形成新型功能性纳米复合材料. ZHANG研究组[25]第一次用简易一步合成g-C3N4-MnO2三明治状结构的纳米复合材料,该材料可以选择性快速识别水溶液和活体细胞中的谷胱甘肽 (GSH),此外,还可以穿透细胞膜,有着很小的细胞毒性,可用于追踪活体细胞内GSH可视化成像技术.
然而,对不同金属元素掺杂的g-C3N4的对比研究相对较少. 因此,本文采用杂化密度泛函理论计算方法,对比研究了在g-C3N4中分别掺杂Mn、Cu和Au三种金属元素前后g-C3N4的电子态、结构参数、电荷布局分布、能隙等变化规律,最终从理论上预测了一类新型的具有高光催化活性的M/g-C3N4复合物.
1计算方法和模型
本文中所有的计算都在Gaussian 09软件包[26]中完成. 采用杂化密度泛函B3LYP[27-28]对g-C3N4以及不同自旋多重度的M/g-C3N4(M=Mn, Au, Cu)的构型进行全优化. 其中对金属原子采用有效核心势耦合的LANL2DZ基组[29],对其他原子采用6-31G基组. 本文采用簇模型,以-C3N4-为结构单元(如图2所示),由于g-C3N4材料中结构单元是无限周期性扩展的,我们通过逐渐加大聚合度(结构单元的数目)的方法来确定合适g-C3N4的团簇结构来模拟材料的周期性结构,并且采用氢原子饱和g-C3N4边界原子的悬键来简化处理团簇结构模型. 在逐渐增加聚合度的基础上,当最终优化的包含n聚合度的g-C3N4构型的能隙与上一较小聚合度的能隙差值小于2 kcal/mol时,认为此团簇结构可以近似模拟半导体材料g-C3N4的周期性结构. 最终我们计算得到聚合度为7时的g-C3N4团簇构型是用来模拟材料的最佳结构模型.
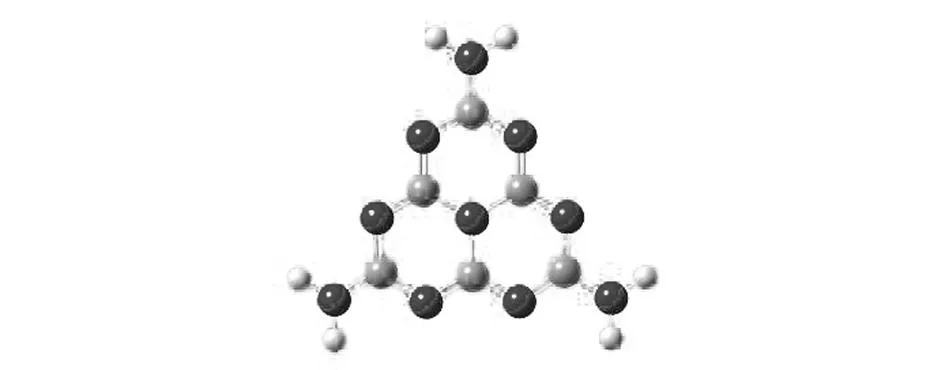
图2 g-C3N4的基本结构单元Fig.2 The unit of g-C3N4
为了比较在g-C3N4中掺杂不同金属原子的难易程度和掺杂后g-C3N4的稳定性,我们分别计算了平均每个金属原子与g-C3N4单元的结合能(Eb),其计算公式如下:
其中EM/g-C3N4为掺杂金属原子后体系的能量,Eg-C3N4为没有掺杂金属原子体系的能量,n 为掺杂的金属原子个数,EM为单个金属原子的能量.
2结果与讨论
2.1g-C3N4的几何结构及电子性质
图2是优化得到的g-C3N4最小结构单元(聚合度为1时的结构). 以这一结构为基础单元,逐渐加大聚合度,在同样的计算水平上分别优化了聚合度为2~15时g-C3N4的结构,优化结果如图3所示. 计算得到每一种聚合度对应的g-C3N4的最高占据轨道(HOMO)、最低空轨道(LUMO)以及HOMO-LUMO能隙(EGap)表1.
从表1数据可知,当聚合度为1的时候,g-C3N4的EGap最大,与半导体材料g-C3N4的带隙理论值2.7eV[30]相差较远,随着聚合度的增大,EGap逐渐降低,当聚合度为7时,g-C3N4的EGap值由4.31eV降低到3.29eV,并且Gap变化趋于平缓,说明此时g-C3N4模型结构正慢慢接近真实材料的结构. 从理论上讲,随着聚合度的无限增大,构建的g-C3N4模型结构越接近材料的周期性结构,计算结果表明,当聚合度由7增大到15时,g-C3N4的Gap值从3.29eV降低至3.17eV,变化并不明显.

表1 聚合度为1~15的g-C3N4优化结构的能隙EGap值
此外,考虑到g-C3N4聚合度为6和7时,其结构有两种排列方式,即三角形排列如图4(a、c)和线形排列如图4(b、d)所示. 我们分别对这两类构型进行优化,结果表明以线形方式排列的结构比较稳定,计算得到的能量和EGap列于表2. 聚合度为6时,这两种结构的能量值相差-35 465.16 kcal/mol,差别非常大,而当聚合度为7时,两种结构的能量值相差25.47 kcal/mol,差别已不是十分明显. 考虑到计算资源等因素,我们取聚合度为7时的g-C3N4结构模型来模拟g-C3N4材料的周期性结构,如图5所示. 此时g-C3N4的Gap值为3.29 eV,接近真实半导体g-C3N4材料2.7 eV的Gap值,计算值与真实值有差异,可能原因是计算所用的方法基组不够精确,不能完全真实描述体系的多电子相互作能;构建的g-C3N4模型聚合度不是足够大,不能模拟真实材料结构;抽取单层的g-C3N4并做简化处理,没有考虑真实材料结构中层与层之间的相互作用等. 本实验是研究掺杂不同金属元素前后g-C3N4材料的Gap、电荷分布、键长键角等变化,掺杂前后所使用的基组方法是相同的,并不影响本实验对掺杂前后体系性质变化规律的分析.
2.2M/g-C3N4(M=Mn, Au, Cu) 的几何结构和电子性质
在优化得到的聚合度为7的g-C3N4结构的基础上,分别在最小结构单元环与环之间的空隙中掺杂Mn、Cu和Au三种金属元素. 优化了得到的M/g-C3N4(M=Mn, Au, Cu)的结构如图6所示,计算得到的能隙和结合能(Eb)列于表3.

图3 3-1-3-15分别为聚合度为2~15时优化后的g-C3N4结构Fig.3 Optimized geometries of g-C3N4 with the different degrees of polymerization

图4 聚合度分别为6、7时g-C3N4的两种不同构型Fig.4 Structures of g-C3N4 with degrees of polymerization of 6 and 7

图5 模拟g-C3N4材料的结构Fig.5 Simulating structure of g-C3N4 material

聚合度图LUMO/a.u.HOMO/a.u.EGap/eV6a-0.09082-0.214433.36b-0.08437-0.205523.307c-0.08763-0.207893.27d-0.08991-0.216053.29

图6 e、f、g分别是聚合度为7时M/g-C3N4 (M=Mn, Au, Cu)优化得到的结构Fig. 6 Optimized structures of M/g-C3N4 (M=Mn, Au, Cu) with degrees of polymerization to 7

物质LUMO/a.u.HOMO/a.u.EGap/eVEb/eV7-0.08991-0.216053.29…Mn/g-C3N4-0.09106-0.129761.05-7.49Cu/g-C3N4-0.09529-0.17372.13-2.82Au/g-C3N4-0.09814-0.16591.84-0.44
由图6可以看出,掺杂金属之后的M/g-C3N4(M=Mn, Cu, Au)仍然保持平面构型,由表3可以看出, M/g-C3N4(M=Mn, Cu, Au)的能隙分别为1.05、2.13、1.84 eV,都比没有掺杂金属元素的g-C3N4的能隙要小得多,都能很好的吸收可见光,其中Mn/g-C3N4的能隙减小最为明显,为1.05 eV,其光谱吸收阈值能很好的扩展至红外范围. 此外,M/g-C3N4(M=Mn, Cu, Au) 的结合能分别为-7.49、-2.82和 -0.44 eV,它们的结合能都为负值,说明在g-C3N4中掺杂Mn、Cu和Au原子能量都是有利的,结构能稳定存在. 综上所述,在g-C3N4中掺杂Mn, Cu, Au原子能极大的增强了g-C3N4的可见光吸收范围,提高了其在可见光条件下的光催化效率. 因此,以下将以Cu/g-C3N4为代表,对其结构参数和电荷布局数进行详细的分析和讨论.
2.3Cu/g-C3N4键长及电荷分析
如图7和8所示,分别是掺杂金属Cu元素前后g-C3N4和Cu/g-C3N4优化之后的结构,掺杂其他金属优化的结构也类似,具体的相应原子上的键长数据见表4.

图7 g-C3N4结构的原子编号Fig.7 Atomic numbers of g-C3N4

图8 Cu/g-C3N4结构的原子编号Fig.8 Atomic numbers of Cu/g-C3N4

键g-C3N4Cu/g-C3N4Mn/g-C3N4Au/g-C3N461N-65C0.14710.14510.14600.146461N-57C0.14730.14690.14340.150561N-101C0.14680.14570.14390.146157C-59N0.13410.13640.13760.139057C-60N0.13450.13530.13830.134556C-60N0.13400.13360.13340.132956C-54N0.14020.14000.13840.139756C-55N0.13280.13280.13200.134558C-50N0.13240.13230.13260.132058C-54N0.14030.13870.13780.138648C-50N0.13360.13550.13740.134049N-48C0.13670.13650.13610.137149N-52C0.13520.13500.13460.135358C-59N0.13380.13310.13260.135051C-55N0.13380.13620.13830.136051C-53N0.13680.13790.13740.138151C-62N0.13980.14090.13880.142352C-53N0.13500.13360.13300.134052C-54N0.14290.14450.14600.144765C-66N0.13370.13530.13720.135269C-66N0.13330.13140.13190.1320
由表4可以看出,掺杂金属前,g-C3N4中61N-57C键长为0.147 3 nm,61N-65C键长为0.147 1 nm,61N-101C键长为0.146 8 nm,这三根键都是C-N单键,说明g-C3N4结构中,基本结构单元环与环之间是通过C-N单键相连形成无限扩展的平面. 此外,54N-56C键长为0.140 2 nm、54N-52C键长为0.142 9 nm、54N-58C键长为0.140 3 nm,这3根键的长度介于单键和双键之间;57C-60N、57C-59N、56C-60N、58C-59N、56C-55N、51C-55N、58C-50N、48C-50N、53N-51C、52C-53N、49N-48C、49N-52C的键长都在0.132 4~0.136 8 nm范围内,为C=N双键,说明g-C3N4基本结构单元环中存在着电子离域效应,形成共轭大π键,共轭π键的存在使得该结构更加稳定,这与KROKE等[18]所得出的3-s-三嗪结构的 g-C3N4更加稳定这一研究结果相符合. 掺杂金属元素后,M/g-C3N4(M=Mn, Cu, Au)的键长与g-C3N4相比没有发生明显的变化,说明掺杂金属元素使g-C3N4原来的构型没有发生太大的改变,仍然保持平面构型.
g-C3N4和Cu/g-C3N4优化得到的构型中的原子的编号如图7、图8所示,掺杂其他金属的结构也同Cu/g-C3N4的结构相类似. M/g-C3N4(M=Mn, Cu, Au) 相应原子上的Mulliken电荷布居数据见表5.

表5 g-C3N4 和M/g-C3N4 (M=Mn, Cu, Au)结构单元中部分原子的Mulliken电荷布局数
由图7、图8和表5可以看出,N原子掺杂金属前后都带负电荷. 掺杂的金属Mn、Cu、Au的电荷布局数分别为0.99 |e|、0.36 |e|和0.42 |e|,都带正电荷. 掺杂金属Cu之后,g-C3N4上的33N和61N上的负电荷由-0.75 |e|变为-0.76 |e|,40N和49N上的电荷由-0.39 |e|变为-0.40 |e|,54N的电荷由-0.73 |e|变为-0.77 |e|,63N由-0.71 |e|变为-0.72 |e|. 最大的变化发生在50N、59N和103N上,其电荷分别由-0.27 |e|、-0.29 |e|和-0.30 |e|变为-0.42 |e|、-0.43 |e|和-0.43 |e|. 同时,金属Mn和Au掺杂之后的变化趋势和Cu掺杂之后的电荷变化是类似的,这些数据表明掺杂金属元素后,电子从金属上转移至环周围的N原子上.
3结论
g-C3N4作为目前光催化研究的热点,因其在环境污染治理、清洁能源再生等方面的应用使其在光催化领域具有很广阔的应用前景. 本文通过向g-C3N4中掺杂不同的金属元素Mn、Cu和Au进一步调节g-C3N4的光催化性能. 结果表明:在g-C3N4中掺杂Mn、Cu和Au原子都是能量有利的,且掺杂后的复合物仍然保持平面构型. 更重要的是它们的能隙都明显减小,能有效的吸收可见光,其中Mn的掺杂甚至能扩展至红外区,这使得g-C3N4的可见红外光催化效率得到明显提高,这也使其可以成为良好的可见红外光催化剂.
参考文献:
[1] FUJISHIMA A, HONDA K. Photolysis-decomposition of water at the surface of an irradiated semiconductor [J]. Nature, 1972, 238(5385): 37-38.
[2] TAKAHASHI H. Status and plans from Hemlock semiconductors [C]. 1st Solar Silicon Conference, Munich, 2004.
[3] 马文会,戴永年,杨斌,等. 加快太阳能级硅制备新技术研发促进硅资源可持续发展[J]. 中国工程科学, 2005(S): 91-94.
[4] 陈威, 王慧, 杨俞, 等. 光催化完全分解水制氢研究进展[J]. 化学研究, 2014, 25(2): 201-208.
[5] 种瑞峰, 吴文鹏, 张凌, 等. 基于二氧化钛光催化剂生物质转化制备氢气和化学品的研究进展[J]. 化学研究, 2015, 26(1): 8-18.
[6] 郭建辉, 杨建军, 李庆霖, 等. 纳米薄膜光催化剂的制备与表面态研究[J]. 化学研究, 2003, 14(2): 13-17.
[7] LIU A Y, COHEN M L. Prediction of new low compressibility solids [J]. Science, 1989, 245(4920): 841-842.
[8] TETER D M, HEMLEY R J. Low-compressibility carbon nitrides [J]. Science, 1996, 271: 53-55.
[9] KROKE E, SCHWARZ M, HORATH-BORDON E, et al. Tri-s-triazine derivatives. Part I. From trichloro-tri-s-triazine to graphitic C3N4structures [J]. New J Chem, 2002, 26(5): 508-512.
[10] WANG X,MAEDA K,THOMAS A,et al. A metal-free polymericphotocatalyst for hydrogen production from water under visible light [J]. Nat Mater, 2009, 8(1): 76-80.
[11] BYERS J C, TAMIASSO-MARTINHON P, DESLOUIS C, et al. Atomic force microscopy studies of carbon nitride (CNx) films deposited on a conducting polymer substrate [J]. J Phys Chem C, 2010, 114(43): 18474-18480.
[12] (a) HAQUE E, JUN J W, TALAPANENI S N, et al. Superior adsorption capacity of mesoporous carbon nitride with basic CN framework for phenol [J]. J Mater Chem, 2010, 20: 10801-10803. (b) ZHOU Z B, CUI R Q, PANG Q J, et al. Schottky solar cells with amorphous carbon nitride thin films prepared by ion beam sputtering technique [J]. Sol Energy Mater Sol Cells, 2002, 70: 487-493.
[13] VINU A. Two-dimensional hexagonally-ordered mesoporous carbon nitrides with tunable pore diameter, surface area and nitrogen content [J]. Adv Funct Mater, 2008, 18: 816-827.
[14] GOETTMANN F, THOMAS A, ANTONIETTI M, et al. Metal-free activation of CO2by mesoporous graphitic carbon nitride [J]. Angew Chem Int Ed, 2007, 46: 2717-2720.
[15] MEEK G A, BACZEWSKI A D, LITTLE D J, et al. Polaronic relaxation by three-electron bond formation in graphitic carbon nitrides [J]. J Phys Chem C, 2014, 118: 4023-4032.
[16] STOLBOV S, ZULUAGA S. Sulfur doping effects on the electronic and geometric structures of graphitic carbon nitride photocatalyst: insights from first principles [J]. J Phys: Condens Matter, 2013, 25: 85507-85513.
[17] HONG J D, XIA X Y, WANG Y S, et al. Mesoporous carbon nitride withinsitusulfur doping for enhanced photocatalytic hydrogen evolution from water under visible light [J]. J Mater Chem, 2012, 22: 15006-15012.
[18] DONG G H, ZHAO K, ZHANG L Z. Carbon self-doping induced high electronic conductivity and photoreactivity of g-C3N4[J]. Chem Commun, 2012, 48: 6178-6180.
[19] LIU G, NIU P, SUN C H, et al. Unique electronic structure induced high photoreactivity of sulfur-doped graphitic C3N4[J]. J Am Chem Soc, 2010, 132: 11642-11648.
[20] CHEN G, GAO S P. Structure and electronic structure of S-doped graphitic C3N4investigated by density functional theory [J]. Chin Phys B, 2012, 21 (10): 107101.
[21] YAN S C, LI Z S, ZOU Z G. Photodegradation of rhodamine B and methyl rrange over boron-doped g-C3N4under visible light irradiation [J]. Langmuir, 2010, 26(6): 3894-3901.
[22] 阮林伟, 裘灵光, 朱玉俊, 等. g-C3N4碳位掺杂电学及光学性质的分析[J]. 物理化学学报, 2014, 30(1): 43-52.
[23] 金瑞瑞, 游继光, 张倩, 等. Fe掺杂g-C3N4的制备及其可见光催化性能[J]. 物理化学学报, 2014, 30(9): 1706-1712.
[24] YE S, QIU L G, YUAN Y P, et al. Facile fabrication of magnetically separable graphitic carbon nitride photocatalysts with enhanced photocatalytic activity under visible light [J]. J Mater Chem A, 2013, 1(9): 3008-3015.
[25] ZHANG X L, ZHENG C, GUO S S, et al. Turn-on fluorescence sensor for intracellular imaging of glutathione using g-C3N4nanosheet-MnO2sandwich nanocomposite [J]. J Anal Chem, 2014, 86: 3426-3433.
[26] FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 09 [CP]. Revision B.01, Wallingford CT: Gaussian, Inc., 2009.
[27] LEE C, YANG W, PARR R G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density [J]. Phys Rev B, 1988, 37: 785-789.
[28] BECKE A D. Density-functional thermochemistry. III. The role of exact exchange [J]. J Chem Phys, 1993, 98: 5648-5652.
[29] COUTY M, HALL M B. Basis sets for transition metals: optimized outer p functions [J]. J Comput Chem, 1996, 17: 1359-1370.
[30] CAO S, YU J. g-C3N4-based photocatalysts for hydrogen generation [J]. J Phys Chem Lett, 2014, 5: 2101-2107.
[责任编辑:吴文鹏]
Theoretical investigation on g-C3N4doped by the different metal atoms
LI Peng, WANG Haiyan*, ZHU Chun*
(SchoolofChemistryandChemicalEngineering,GuizhouUniversity,Guiyang550025,Guizhou,China)
Abstract:The geometries of g-C3N(4 )doped by M (M=Mn, Cu, Au) have been optimized, and their energy gaps, binding energies, frontier orbitals have been characterized by density functional theory calculations. The results demonstrate that the stable order is Mn/g-C3N4>Cu/g-C3N4>Au/g-C3N4. Furthermore, metal doping can significantly reduce the energy gap of g-C3N4, increase its absorption in visible region and improve its photocatalytic efficiency. The research can provide the theoretical guidance to understand the effection of modification on the photocatalytic efficiency of g-C3N4.
Keywords:density functional theory; photocatalysis meterials; g-C3N4; metal doping; energy gap
文章编号:1008-1011(2016)02-0152-09
中图分类号:O641.12
文献标志码:A
作者简介:李鹏(1990-),男,硕士生,研究方向为理论与计算化学. *通讯联系人, E-mail: sci.hwang@gzu.edu.cn; czhu2014@163.com.
基金项目:教育部春晖计划 (Z2014088),贵州省科学技术基金(【2012】2151).
收稿日期:2016-02-18.
猜你喜欢
首都师范大学学报(自然科学版)(2022年5期)2022-10-19
黑龙江大学工程学报(2022年1期)2022-04-06
云南民族大学学报(自然科学版)(2022年1期)2022-02-12
物理学报(2021年12期)2021-07-01
数学物理学报(2020年6期)2021-01-14
中国材料进展(2017年9期)2017-10-13
科技资讯(2016年5期)2016-08-13
武汉工程大学学报(2016年1期)2016-04-07
原子与分子物理学报(2015年2期)2015-03-23
浙江师范大学学报(自然科学版)(2013年2期)2013-10-25
