
共缩聚反应中链段序列长度调控的分子模拟研究
2016-07-10 21:07张怀哲刘静马禹余木火
当代化工 2016年4期
张怀哲 刘静 马禹 余木火


摘 要:利用分子模拟研究了共缩聚反应得到的高分子链上的嵌段长度分布。通过控制参加反应的两种组分的浓度比和共聚单体的反应活性,可得到与理论值差别很大的序列分布。有助于合成嵌段长度可控的共聚高分子。
关 键 词:分子模拟;共缩聚;嵌段长度
中图分类号:TQ 325 文献标识码: A 文章编号: 1671-0460(2016)04-0724-03
Abstract: In this study, molecular simulation was employed to study the block length distribution along polymer chain obtained by cocondensation polymerization. Its found that the distribution exhibits a significant deviation from the theoretical predictions, particularly for two components with different concentration and reactivity. This research is helpful for synthesis of copolymer with controlled block length.
Key words: Molecular simulation; Cocondensation polymerization; Block length distribution
化学改性高分子材料的方法有很多种,对缩聚制备的高分子材料而言,在聚合物主链上通过共缩聚引入共聚单体,控制共聚单元的序列长度,是最常用的方法。利用分子模拟的手段对共缩聚反应进行研究,反应条件可以准确控制,实验结果重复性好,有助于高效快速地筛选出合适的改性方案,是对实验和理论研究很好的补充[1]。本文基于该模拟方法,首次对共缩聚反应进行了研究,着重考察了无热体系中,共聚单体浓度和竞聚率对其在聚合物主链上分布的影响。
1 模拟方法和理论分析
反应模拟在边长为64的三维立方格子空间中进行,利用周期性边界条件消除有限元胞效应。单体和高分子链上的链单元可以通过单格点跳跃的方式在格子空间中运动,运动过程不能打断分子链或违背体积排除原则。该模拟方法可以得到符合真实高分子单链或多链标度关系的构象和链动力学信息。这种运动模型已经被广泛地用于单链以及多链体系的相变、转变及多级结构的形成机理研究[2,3]。我们定义所有链单元进行一次尝试运动所需的抽样次数为一个蒙特卡罗循环(MCC),等效于反应时间t。在每个循环中,所有单体和链端基进行一次尝试反应,即链端基可以和位置邻近的另一个链的端基反应,形成新的键把两个分子连接成一个新的分子。该尝试反应被接受的几率定义为k,物理意义类似于实际反应中的反应速率常数。随着反应的进行和新键的形成,单体逐渐消失,分子数目逐渐减少,最终得到高分子量聚合物。在本研究中,我们不考虑除体积排除外的一切相互作用,反应中不允许发生环化、链交换和解聚。
反应初始时,加入浓度分别为ΦA、ΦB的A和B两种单体,单体的总浓度为5%。分别以kAA、kBB、kAB、kBA表示单体A与单体A、单体B与单体B、单体A与单体B以及单体B与单体A之间反应接受的几率,且满足kAB=kBA。反应几率仅取决于链端单元的种类,而与较远的链单元无关。类似自由基共聚反应,定义竞聚率rA=kAA/kAB,rB=kBB/kBA,表示链单元自身结合与交叉结合的动力学差异。在模拟中,设定kBB=kAB=kBA=0.1,通过改变kAA的值,可以获得不同竞聚率的反应条件。
基于概率分析[4],可以得到出现长度为l的A嵌段的几率PA(l)和B嵌段的几率PB(l)分别为:
积分(1)(2)式可得链段A和B的平均长度分别为:
上述的理论分析中,实际上隐含了链单元均匀分布、链端单元出现几率恒定以及无限链长等一系列严格的假定,与真实反应相比可能过于理想。而且理论分布只解释了反应最终的序列分布,而无法给出反应过程中链段序列长度随反应时间的变化。因此,采用分子模拟的方法,在共聚反应中,通过加入不同浓度和不同竞聚率的共聚单体,对实际嵌段长度分布与理论值的偏差进行研究。
2 结果及讨论
2.1 ΦA:ΦB=0.5:0.5的情形
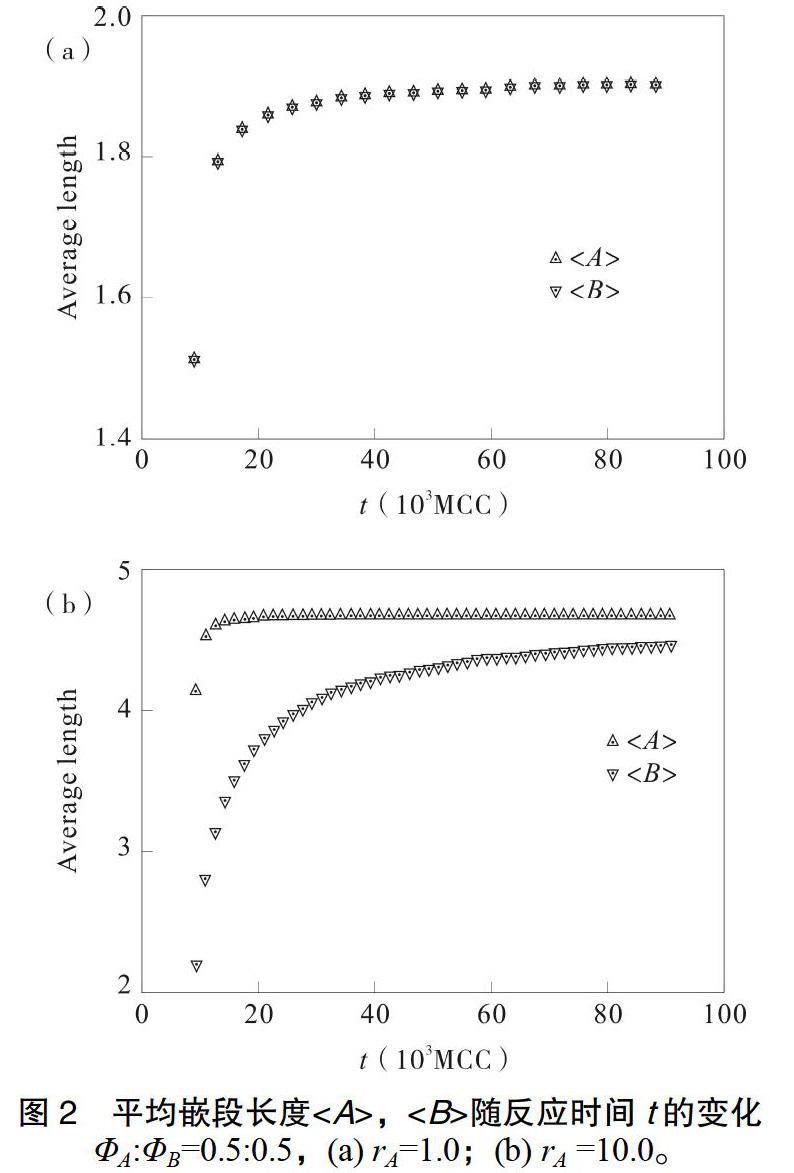
在无热反应中,等摩尔比投入A和B两组分,kAA分别设为0.01、0.1、1.0,从而可得rA分别为0.1、1.0、10.0,rB为1.0的反应体系。统计结果取自反应程度超过0.99的状态。
从表1和图1中可以看到,当rA=1.0时,模拟结果与理论预测符合程度较高;而当rA分别为0.1和10.0时,偏差较大。尤其是rA为10.0时,A嵌段短序列出现几率明显高于理论值,而长序列则显著偏低,导致A嵌段平均序列长度仅为理论值一半。理论分布是建立在不同种类链端基消耗速率恒定的基础上推导的,而该假定实际上仅对rA=rB=1时成立。以rA=10.0为例,如图2(b),由于A组分反应速率较快,在反应初期A单体即迅速消耗完毕,形成了以B组分封端的低聚体。随着反应的进一步进行,低聚体间两两结合,B嵌段的平均长度随反应进行而逐渐增加。
由于A与B两组分单体在早期浓度高且分布均匀,在较高的交叉反应速率(kAB)下,无法直接得到长的A嵌段,而后期反应中,B嵌段一方面在链端进一步生长,一方面不同分子端部的B嵌段两两合并,最终导致B嵌段序列长度比理论值高一倍,而A嵌段长度不到理论值的一半。
此外,由表1可以看到,即使添加的共聚单体反应活性差别显著,也不会明显改变A和B两嵌段的长度比。长度比更多地由反应的投料比所控制而不是反应活性,这也是逐步聚合共聚和自由基聚合共聚的显著差别。同时,加入的共聚单体反应活性提高,则两种嵌段序列长度同时增加;反之则同时减少,可以利用控制单体反应活性的方法制备序列长度可调的多嵌段聚合物。
2.2 ΦA:ΦB=0.05:0.95的情形
不等摩尔比投料常用于对现有高分子的结构和性能进行细微改变,往往仅加入极少量的共聚单体。在模拟中,加入A和B两组分比例为0.05:0.95其他反应条件同前。
由表2可知,与等摩尔比投料相比,添加少量A组分不会带来与理论预测值的显著差别,这主要因为A组分含量较少,形成长的A嵌段可能性较小。在最终所得的聚合物中,A组分绝大多数以游离的形式插入在分子链上。B嵌段长度受A组分的反应活性影响较大,尤其当A组分反应活性较高时(rA=10.0),A嵌段在反应早期被迅速消耗掉。在早期形成的低聚物中,主要为B组分的均聚物或以B组分封端的低聚物。因此,直至反应后期,B嵌段长度还能够进一步地增加,如图3(b)。
我们的模拟结果说明,当需要通过共聚的方式破坏主链规整性时,应考虑共聚组分的反应活性。只有加入的共聚单体反应活性不低于基体单体时,才能够实现添加最少的组分,同时保证有效控制序列长度。
3 结 论
本文通过分子模拟的方法研究了反应单体浓度比和反应单体活性对共聚物嵌段序列长度的影响。我们发现,在等摩尔比投料情况下,当加入反应活性较高的共聚单体时,两种链段的平均长度均增加,可以实现多嵌段聚合物的制备;而当少量共聚时,只有加入反应活性较低的共聚单体,才能有效控制主链上的序列长度。
参考文献:
[1] 李春艳,刘华,刘波涛.分子模拟的方法及其应用[J].当代化工,2011(05):494-497.
[2] Yu Ma, WenbingHu, Günter Reiter. Lamellar crystal orientations biased by crystallization kinetics in polymer thin films[J]. Macromolecules, 2006, 39(15): 5159–5164.
[3] Yu Ma, Liyun Zha, Wenbing Hu, Günter Reiter, Charles C. Han. Crystal nucleation enhanced at the diffuse interface of immiscible polymer blends[J]. Phys Rev E, 2008, 77:061801.
[4] 唐敖庆, 等.高分子反应统计理论[M].北京:科学出版社, 1985: 293-324.
猜你喜欢
江西水产科技(2022年2期)2022-05-17
当代化工(2019年3期)2019-12-12
三联生活周刊(2017年48期)2017-11-25
课堂内外·教师版(2017年3期)2017-04-13
科技视界(2016年7期)2016-04-01
党的生活(黑龙江)(2015年7期)2015-07-14
中国民族民间医药·下半月(2014年4期)2014-09-26
时代英语·高三(2014年5期)2014-08-26
哈尔滨理工大学学报(2014年1期)2014-06-23
中学生数理化·高二版(2008年6期)2008-11-12
