
Structural Effect on Absorption and Emission Properties of 1,8-Naphthalimide Derivatives: a DFT Study
2016-07-12 12:49QIQiWANGYuqiaoSUNYueming
光谱学与光谱分析 2016年11期
QI Qi, WANG Yu-qiao, SUN Yue-ming
1. School of Chemistry and Chemical Engineering, Southeast University, Nanjing 211189, China 2. Jiangsu Optoelectronic Functional Materials and Engineering Laboratory, Nanjing 211189, China 3. Beijing Computational Science Research Center, Beijing 100084, China
Structural Effect on Absorption and Emission Properties of 1,8-Naphthalimide Derivatives: a DFT Study
QI Qi1,2,3, WANG Yu-qiao1,2*, SUN Yue-ming1,2*
1. School of Chemistry and Chemical Engineering, Southeast University, Nanjing 211189, China 2. Jiangsu Optoelectronic Functional Materials and Engineering Laboratory, Nanjing 211189, China 3. Beijing Computational Science Research Center, Beijing 100084, China

1,8-naphthalimide derivatives; Frontier orbitals; Absorption spectra; Emission spectra; Density function theory
Introduction
1,8-naphthalimide derivatives (NI, Fig.1) recently have been widely utilized as dyes and luminescent materials because of chromophores’ properties in visible light region. High electron affinity[1], wide gap[2-3], and low reduction potential[4], make them good candidates as n-type materials[5-7]. 1,8-naphthalimide derivatives also found applications in sensitizers of Grätzel solar cell[8-9], logic gates[10], sensors[11-13]and charge separation mimics[14], where they could act as either acceptors or donors of electron and energy[15-22]. Besides, 1,8-naphthalimides have also been used as fluorescent markers for the study of dynamic protein interactions and phosphorescent organic LED[23].

Fig.1 Scheme picture of the 1,8-naphthalimide derivatives. (Me- methyl; Ph- phenyl) (B1 and bond indexes stand for bond interaction between R4 or R5 and naphthalimide ring)
Above applications thus push forward the development and design of 1,8-naphthalimides molecular engineering. 1,8-naphthalimides typically absorb at 350~450 nm region and emit at a wavelength roughly longer by 100 nm[3,24-25]. Their excited state can be tuned by the nature of a substituent present on the aromatic ring so the colorless compounds and weak fluorescent can be improved[25]. In the presence of substituent, they become yellow, which also induces a polar charge transfer excited state. Semi-empirical models and TDDFT methods were employed to investigate the geometrical and electronic structures of these molecules[26-32]. B3LYP with a 6-31+G(d) basis set including solvent effect were verified to be in good agreement with experimental spectra by Ksenija and Denis[26]. They illustrate imide seldom participate the contribution of the frontier orbital and imide moiety plays a marginal role in the emission spectra, which is mainly related to a modification of the C—C π-bonding pattern. On the other hand, 4-position substituents of modified 1,8-naphthalimides with diphenylamino, dimethylamino, amino, and nitro[33-36]units also show broad absorptions around 450~480 nm. Modification of imide group and asymmetry of the whole NI unit is involved in structural changes. Compare with original imide, some modified derivatives shows orange color from yellow color[32]. They also exhibit their intermolecular π—π*charge transfer properties[33].
In this paper, our aim is to investigate the modified 1,8-naphthalimides (Fig.1), as well as their absorption and emission spectra in gas-phase and dichloromethane. Our focus is on the structural modification of imide groups and 1,8-naphthalimides’r spectral characteristic. The structure and excited state properties in solvent of 1,8-naphthalimides are also investigated to provide further reference for these kinds of molecules. Their charge transfer state properties and their energy gap are also analyzed.
1 Computation Methods
All calculations have been carried out with the Gaussian 09 package[34]. We have chosen the B3LYP[35-36]functional for all the DFT and TD-DFT computations. 6-31+G(d) basis set has been used to obtain the excitation properties based on 1,8-naphthalimides’ optimized structures. In order to compute the absorption and fluorescent spectra, the molecular geometries of the ground state (S0) and the first singlet excited state (S1) have been optimized with HF (S0) and CIS (S1) approaches, respectively. Natural orbital population[37-38]and natural bond orbital[39]analyses were carried out to compute dipole moments and bond indexes. The optimized S0 and S1 structures were subsequently used to compute the absorption and the emission spectra with the TDDFT method, where the solvent effect of dichloromethane is taken into consideration with the C-PCM[40-41]model.
2 Result and discussion
2.1 Geometries of 1,8-naphthalimide derivatives in the S0 and S1 states

Table 1 Partial selected bond lengths [nm], NBO bond indices [a.u.], dipole moments [a.u.] and mulliken atomic charges of substituents(MAC) in ground state and excited state calculated at HF/6-31G*, CIS/6-31G*
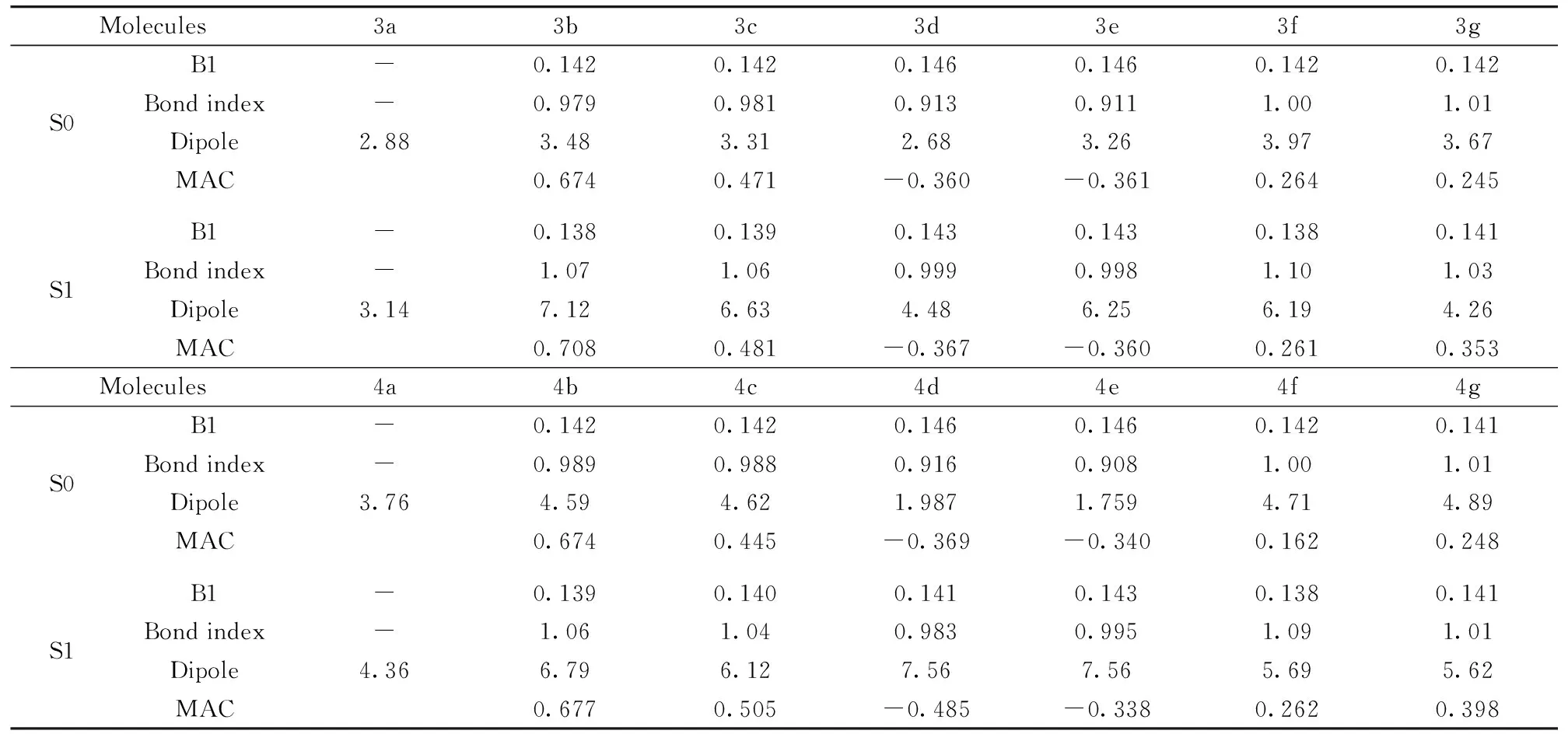
Molecules3a3b3c3d3e3f3gS0B1-0.1420.1420.1460.1460.1420.142Bondindex-0.9790.9810.9130.9111.001.01Dipole2.883.483.312.683.263.973.67MAC0.6740.471-0.360-0.3610.2640.245S1B1-0.1380.1390.1430.1430.1380.141Bondindex-1.071.060.9990.9981.101.03Dipole3.147.126.634.486.256.194.26MAC0.7080.481-0.367-0.3600.2610.353Molecules4a4b4c4d4e4f4gS0B1-0.1420.1420.1460.1460.1420.141Bondindex-0.9890.9880.9160.9081.001.01Dipole3.764.594.621.9871.7594.714.89MAC0.6740.445-0.369-0.3400.1620.248S1B1-0.1390.1400.1410.1430.1380.141Bondindex-1.061.040.9830.9951.091.01Dipole4.366.796.127.567.565.695.62MAC0.6770.505-0.485-0.3380.2620.398
B1 and Bond indexes stand for the bond length of C—N between R4/R5 substituents and naphthalimide in Fig.1. See Fig.1 for atom numberings
2.2 Frontier molecular orbitals (FMOs) analysis and their energy level analysis




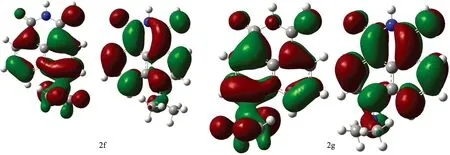


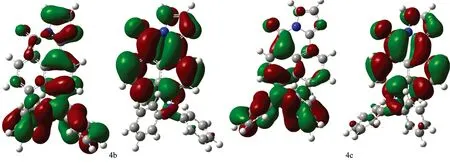
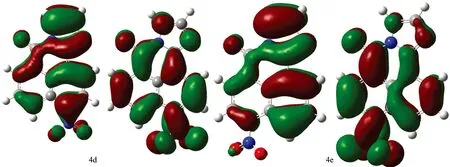
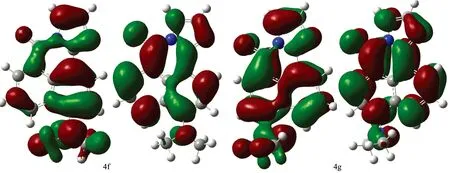
Fig.2 Contour plot for HOMOs, LUMOs of 1, 8-naphthalimide derivatives calculated by b3lyp/6-31+G(d) with the CPCM(ethanol)-B3LYP/6-31+G(d) scheme (The first row show their HOMOs while the second row show their LUMOS) (1a—4g)

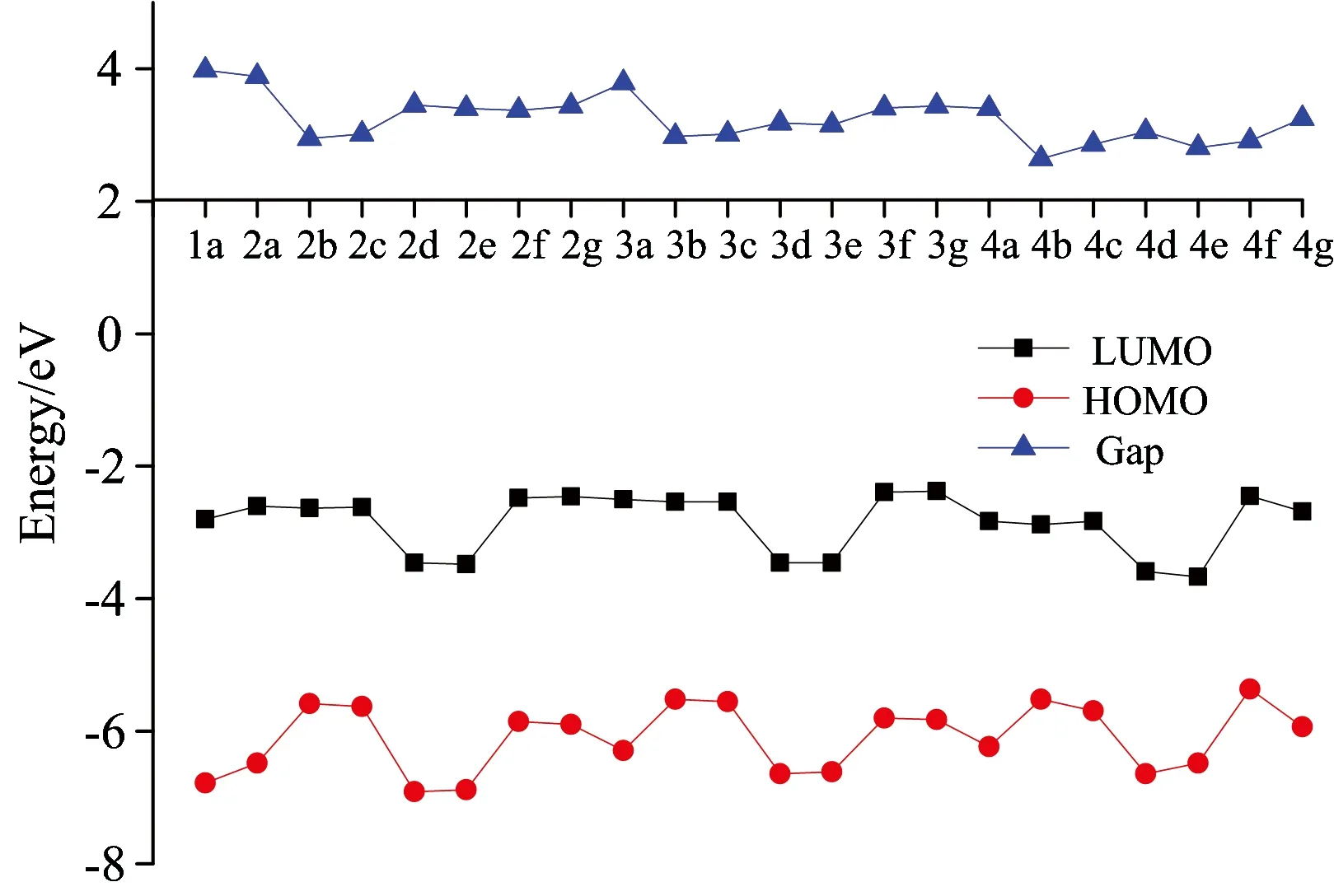
Fig.3 Energy level of HOMO, LUMO, Gap for compounds calculated at B3LYP/6-31+G* level (in eV)
2.3 Absorption, fluorescence properties in gas phase and dichloromethane
The data of vertical excitation energy and oscillator strength in dichloromethane are listed in Table 2. Here major electronic absorption bands are assigned to those excitations with significant oscillator strengths.
Table 2 Vertical absorption and emission energies in gas phase and in dichloromethane calculated at TD-B3LYP/6-31+G* level within C-PCM model. Their absorption wavelengths and energies are listed in unit of nm (eV).λabs(nm)λemis(nm)
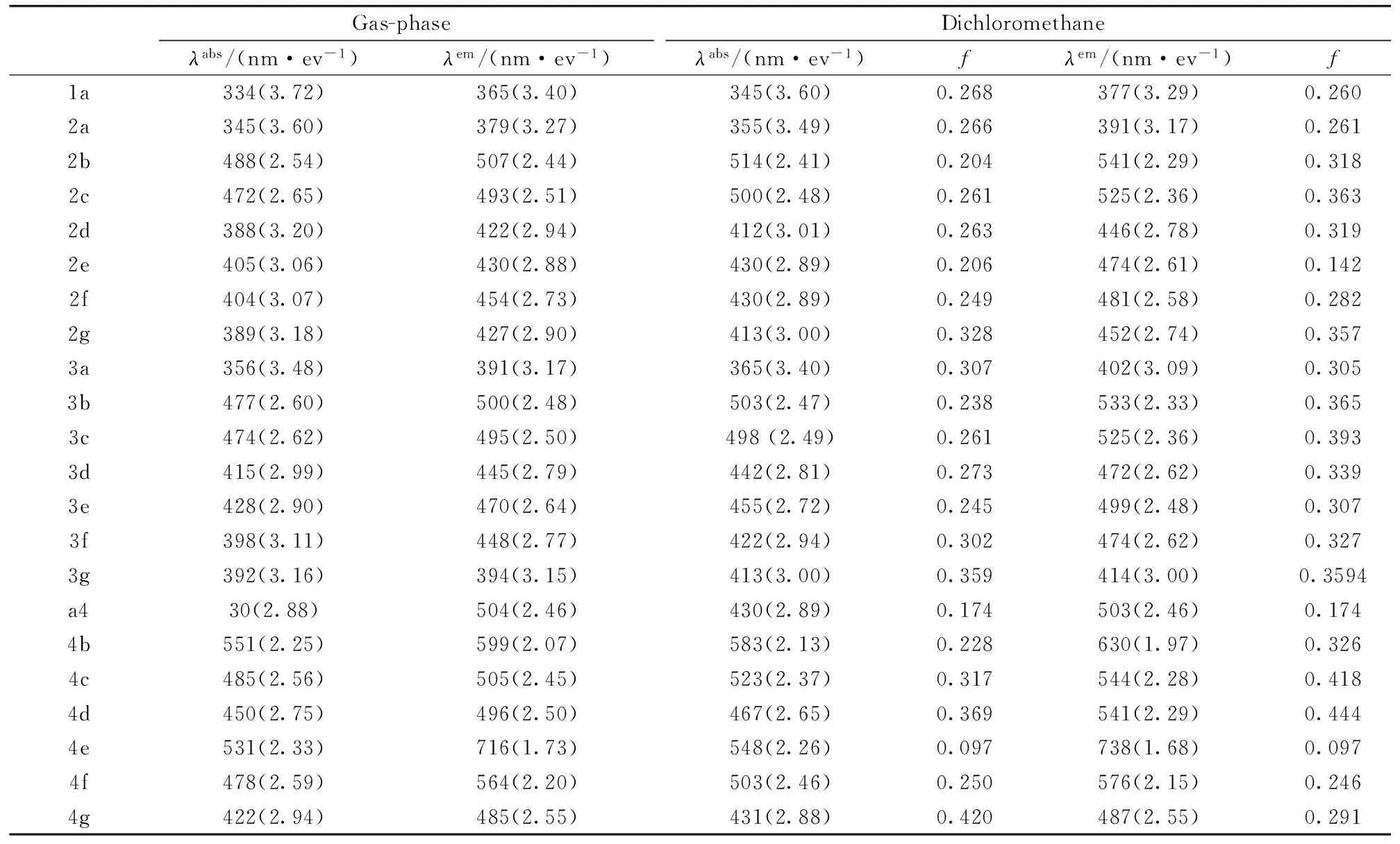
Gas-phaseDichloromethaneλabs/(nm·ev-1)λem/(nm·ev-1)λabs/(nm·ev-1)fλem/(nm·ev-1)f1a334(3.72)365(3.40)345(3.60)0.268377(3.29)0.2602a345(3.60)379(3.27)355(3.49)0.266391(3.17)0.2612b488(2.54)507(2.44)514(2.41)0.204541(2.29)0.3182c472(2.65)493(2.51)500(2.48)0.261525(2.36)0.3632d388(3.20)422(2.94)412(3.01)0.263446(2.78)0.3192e405(3.06)430(2.88)430(2.89)0.206474(2.61)0.1422f404(3.07)454(2.73)430(2.89)0.249481(2.58)0.2822g389(3.18)427(2.90)413(3.00)0.328452(2.74)0.3573a356(3.48)391(3.17)365(3.40)0.307402(3.09)0.3053b477(2.60)500(2.48)503(2.47)0.238533(2.33)0.3653c474(2.62)495(2.50)498(2.49)0.261525(2.36)0.3933d415(2.99)445(2.79)442(2.81)0.273472(2.62)0.3393e428(2.90)470(2.64)455(2.72)0.245499(2.48)0.3073f398(3.11)448(2.77)422(2.94)0.302474(2.62)0.3273g392(3.16)394(3.15)413(3.00)0.359414(3.00)0.3594a430(2.88)504(2.46)430(2.89)0.174503(2.46)0.1744b551(2.25)599(2.07)583(2.13)0.228630(1.97)0.3264c485(2.56)505(2.45)523(2.37)0.317544(2.28)0.4184d450(2.75)496(2.50)467(2.65)0.369541(2.29)0.4444e531(2.33)716(1.73)548(2.26)0.097738(1.68)0.0974f478(2.59)564(2.20)503(2.46)0.250576(2.15)0.2464g422(2.94)485(2.55)431(2.88)0.420487(2.55)0.291
For 1,8-naphthalimides in dichloromethane, calculated absorption and emission properties are in good agreement with experimental absorption maxima and fluorescence maxima of 346 and 378 nm[3]. Solvent effects make calculation results approach more closely to experimental data as illustrated for 1a.

For compounds with electron donation substituents, absorption maxima of the 4 position compounds are bigger than those 5-positions, which can be attributed to their charge transfer properties. For the NO2group, the 5 position is the charge transfer state which can be seen from its longer red-shift.
3 Conclusion
Acknowledgement: Authors also thank Prof. Liu Chungen in School of Chemistry and Chemical Engineering Nanjing University for discussing this paper.
[1] Cacialli F, Friend R H, Bouche C M, et al. J. Appl. Phys.,1998,83(4): 2343.
[2] Demeter A, Berces T, Biczok L, et al. J. Phys. Chem., 1996,100(6): 2001.
[3] Wintgens V, Valat P, Kossanyi J, et al. J. Chem. Soc. Faraday Trans., 1994,90(3): 411.
[4] Martin E, Weigand R. J. Chem. Soc. Faraday Trans., 1998, 288(1): 52.
[5] Kolosov D, Adamovich V, Djurovich P, et al. J. Am. Chem. Soc., 2002, 124(33): 9945.
[6] Herrera H, Echegaray P D, Urdanpilleta M, et al. Chem. Commun., 2013, 49: 713.
[7] Ortiz R P, Herrera H, Mancheo M J, et al. Chem. Eur. J., 2013, 19(37): 12458.
[8] Zhu W H, Tian H, Gao E Q, et al. Chem. Lett., 2000, 29(7): 778.
[9] Tian H, Liu P H, Zhu W H, et al. J. Mater. Chem., 2000, 10(12): 2708.
[10] Lukas A S, Bushard P J, Wasielewski M R, et al. J. Am. Chem. Soc., 2001, 123(10): 2440.
[11] Elbert J E, Paulsen S, Robinson L, et al. J. Photochem. Photobiol. A, 2005, 169(1): 9.
[12] Georgiev N I, Asiri A M, Alamry K A, et al. J. Photochem. Photobiol. A, 2014, 277(1): 62.
[13] Wang M, Xu Z, Wang X, et al. Dyes Pigments.,2013,96(2): 333.
[14] Le T P, Rogers J E, Kelly L A, et al. J. Phys. Chem. A, 2000,104(29): 6778.
[15] Tan Q, Kuciauskas D, Lin S, et al. J. Phys. Chem. B, 1997,101(26): 5214.
[16] Cho D W, Fujitsuka M, Sugimoto A, et al. J. Phys. Chem. B, 2006,110(23): 11062.
[17] Kitchen J A, Martinho P N, Morgan G G, et al. Dalton Trans., 2014, 43(17): 6468.
[18] Ledwon P, Brzeczek A, Pluczyk S, et al. Electrochimica Acta,2014,128(10): 420.
[19] Plyusnin V F, Kupryakov A S, Grivin V P, et al. Photoch Photobio Sci.,2013,9(12): 1666.
[20] Pogozhev D V, Bezdek M J, Schauer P A, et al. Photochem Photobio Sci.,2013,52(6): 3001.
[21] Abad S, Kluciar M, Miranda M A, et al. J. Org. Chem., 2005, (25): 10565.
[22] Loving G, Imperiali B. J. Am. Chem. Soc., 2008,130(41): 13630.
[23] Kolosov D, Adamovich V, Djurovich P, et al. J. Am. Chem. Soc., 2008, 124(33): 9945.
[24] Alexiou M S, Tychopoulos V, Ghorbanian S,et al. J. Chem. Soc. Perk. Trans., 1990,2(5): 837.
[25] Jacquemin D, Perpète E A, Scalmani G, et al. Chem. Phys. Lett., 2007,448(3): 3.
[26] Miao L, Yao Y, Yang F, et al. J. Mol. Struct: THEOCHEM., 2008,865: 79.
[27] O’Shea Vadp A P, Poyato J M L. Int. J. Quantum. Chem., 2003, 91(3): 446.
[28] Patsenker L D, Artyukhova Y Y. J. Mol. Struct., 2003,655(2): 311.
[29] Jacquemin D, Perpète E A, Scalmani G, et al. Chem. Phys., 2010, 372: 61.
[30] Laurent A D, Adamo C, Jacquemin D. Phys. Chem. Chem. Phys., 2014,16: 14334.
[31] Kucheryavy P, Li G, Vyas S. J. Phys. Chem. A, 2009,113: 6453.
[32] Yang H, Lao Y N, Chen J M, et al. Eur. J. Inorg. Chem., 2009,19: 2817.
[33] Jiang W, Tang J, Qi Q, et al. Dyes Pigments.,2009,80(3): 279.
[34] Frisch M J, Trucks G W, Schlegel H B, et al. Gaussian09, Revision B.01. Walliford, CT: Gaussian, Inc.,2009.
[35] Becke A D. J. Chem. Phys., 1993,98(7): 5648.
[36] Miehlich B, Savin A, Stoll H, et al. Chem. Phys. Lett., 1989,157(3): 200.
[37] Reed A E, Weinstock R B, Weinhold F. J. Chem. Phys., 1985,83(2): 735.
[38] Reed A E, Weinhold F. J. Chem. Phys., 1983,78(6): 4066.
[39] Foster J P, Weinhold F. J. Am. Chem. Soc., 1980,102: 7211.
[40] Barone V, Cossi M. J. Phys. Chem. A, 1998,102: 1995.
[41] Cossi M, Rega N, Scalmani G, et al J. Comput. Chem.,2003,24(6): 669.
O641
A
1,8-萘酰亚胺类衍生物的结构特性对其吸收和发射光谱影响的密度泛函研究
齐 齐1,2,3,王育乔1,2*,孙岳明1,2*
1. 东南大学化学化工学院,江苏 南京 211189 2. 江苏省光电功能材料工程实验室, 江苏 南京 211189 3. 北京计算科学研究中心,北京 100084
1,8-萘酰亚胺;前线轨道;吸收光谱;发射光谱;密度泛函
2015-07-01,
2015-11-20)
Foundation item: the National Natural Science Fund Committee (21173042,21201034), support by the Priority Academic Program Development of Jiangsu Higher Education Institutions, Graduate education reform project (XJGKT14-04), Fundamental Research Funds for the Central Universities (2242014k10025, 3207045419), the financial assistance from Nanjing science and technology committee (2014-030002), the high performance computation platform of Southeast University
10.3964/j.issn.1000-0593(2016)11-3796-09
Received: 2015-07-01; accepted: 2015-11-20
Biography: QI Qi, (1980—), lecture in Southeast University e-mail: qiqi@seu.edu.cn *Corresponding authors e-mail: yqwang@seu.edu.cn; sun@seu.edu.cn
猜你喜欢
东南大学学报(医学版)(2022年1期)2022-03-17
东南大学学报(医学版)(2021年6期)2022-01-27
化工职业技术教育(2021年5期)2021-11-09
东南大学学报(医学版)(2021年5期)2021-11-06
东南大学学报(医学版)(2021年4期)2021-11-05
化工职业技术教育(2021年1期)2021-03-15
民用飞机设计与研究(2020年1期)2020-05-21
化工职业技术教育(2020年4期)2020-01-19
中国塑料(2017年2期)2017-05-17
化工学报(2016年3期)2016-03-14
