
脱氧青蒿素分子光谱性质的密度泛函模拟与指认
2016-07-31 17:08杨星月蒋罗颖陈宇玉尚欣雨
新疆师范大学学报(自然科学版) 2016年2期
龙 威, 杨星月, 蒋罗颖, 陈宇玉, 尚欣雨
(南华大学 化学化工学院,湖南 衡阳 421001)
脱氧青蒿素分子光谱性质的密度泛函模拟与指认
龙 威, 杨星月, 蒋罗颖, 陈宇玉, 尚欣雨
(南华大学 化学化工学院,湖南 衡阳 421001)
文章采用密度泛函理论的DFT/B3LYP方法和6-311+G(d,p)基组,对脱氧青蒿素分子的UV-Vis光谱、ECD光谱、IR光谱、拉曼光谱、1HNMR光谱和荧光光谱进行了理论模拟和指认。Fukui函数扫描得知,2C、8C原子很可能是其发挥药理活性的亲电亲核反应部位,多环之间存在着协同作用可发挥药效。这些可为计算化学方法解释分子结构决定药效发挥、掌握药物分子性质,提供一个科学合理的理论指导。
脱氧青蒿素;密度泛函;电子光谱;量子化学
青蒿素是一种有过氧基团的倍半萜内酯药物,也是继乙氨嘧啶、氯喹、伯喹之后最有效的抗疟特效药,尤其是对于脑型疟疾和抗氯喹疟疾,具有速效和低毒的特点,曾被世界卫生组织称做是“世界上唯一有效的疟疾治疗药物”。2015年10月8日,中国科学家屠呦呦获2015年诺贝尔生理学或医学奖,成为第一个获得诺贝尔自然学奖的中国人。通过多年从事中药和中西药结合研究的科学家屠呦呦,创造性地研制出抗疟新药——青蒿素和双氢青蒿素,获得对疟原虫100%的抑制率,为中医药走向世界指明一条方向[1-2]。
青蒿素也是常见的一种常见的半合成光谱青霉素类药物,其化学主要成分分子式为C15H22O5,CAS为(No.26787-78-0),标准化学名称为(3R,5aS,6R,8aS,9R,12S,12aR)-八氢-3,6,9-三甲基-3,12-桥氧-12H-吡喃[4,3-j]-1,2-苯并二塞-10(3H)-酮[3]。药理上,抗疟疾作用机理主要在于在治疗疟疾的过程中通过青蒿素活化产生自由基,自由基与疟原蛋白结合,作用于疟原虫的膜系结构,使其泡膜、核膜以及质膜均遭到破坏,线粒体肿胀,内外膜脱落,从而对疟原虫的细胞结构及其功能造成破坏[4-7]。我国对青蒿素药物的研究十分精深[8],以屠呦呦为核心的研究团队对数十种青蒿素药物进行临床的试用与研制,对世界医疗卫生事业做出了突出的贡献。世卫组织数据表明,撒哈拉以南非洲地区自2010年以来约2.4亿人口受益于青蒿素联合疗法,约150万人因该疗法避免了疟疾导致的死亡。因此,青蒿素药物被很多非洲民众尊称为“东方神药”。
青蒿素作为一种神奇的分子,已广泛运用在抑制肿瘤的增殖、诱导肿瘤细胞凋亡、转移肿瘤细胞或肿瘤细胞侵袭的抑制等方面,受到了广大医学、药物化学工作者的青睐[9]。汪大婷等[10]使用青蒿素模拟热量限制(CR)延长了酵母的期望寿命,通过实验表明,抗疟药青蒿素易在体内形成青蒿琥酯,并通过烷化血红素蛋白达到青蒿琥酯-血红素结合,从而发挥了类似一氧化氮-血红素相互作用。同时,它与细胞色素c氧化酶活性升高之间有很强的相关性,使用这种药物可以达到抗氧化反应、清除活性氧、降低氧化应激、促进机体由合成代谢向分解代谢转变等重大疗效;盛庆寿等[11]也通过实验表明,双氢青蒿素对原发性肝癌大鼠的治疗是通过抑制大鼠血清VEGF及受体的浓度,抑制了肝癌血管的生成和转移,从而延缓了肿瘤的扩散,达到了肝脏功能的保护;邵义如等[12]发现二氢青蒿素分子对结肠癌细胞系SW480增殖凋亡有明显的药理疗效。因此,青蒿素分子越来越受到科学研究工作者的关注,其独特的性能也逐步被继续挖掘和提高。
大量研究[13-15]表明,药物分子的药效与分子结构特点有一定的联系。笔者[16,17]就曾经使用量子化学方法对几种常见的镇痛药物分子活性的高低进行模拟计算,获得了较好的成果。先进的量子化学计算方法,常用于药物分子的活性分析,从理论上指导实验合成的新方法,往往具有非常积极的科学意义。目前,国内外对青蒿素分子的理论研究尚未报道,我们的研究刚好能有力地补充这一空白,具有积极的意义。
由于高度复杂的青蒿素分子空间结构给我们的量化研究带来困扰,所以我们选择脱氧青蒿素分子作为研究对象,它的分子结构如图1所示,其平面结构如图1(a)所示,立体结构如图1(b)所示,且标有相应的元素符号和原子序号。
分子中含有三个环,分别为两个六元环和一个七元环、同时含O元素,主要有酯基、醚氧基、甲基等官能团,这样特殊的三环联体结构肯定存在着特殊的效应,为了进一步获得相关的信息,以此脱氧青蒿素分子为对象进行了相关的量化研究。
1 计算方法
对脱氧青蒿素分子采用DFT理论的B3LYP方法在6-311+G(d,p)基组水平上进行了优化计算。在优化得到的稳定构型基础上,采用频率分析法,结果表明所有的简谐振动频率全部为正数,表明其计算结果是可信的,本项目的大部分工作通过Gaussian03W程序包[18]在PC机上独立完成,部分计算采用了科学计算网格ScGrid协助完成。
定域反应活性指数应用Fukui函数[19]分别是电子密度ρ(r)对电子数N的一阶微商。因为 f(r)=(ρ(r)/N)ν,由于Fukui函数是非连续性的,运用有限差分近似方法简化亲电Fukui函数、亲核Fukui函数,可以获得:上述式中 ρN(r)、ρN-1(r)、ρN+1(r)分别是中性分子、阳离子和阴离子的电子密度[20]。考虑到分子的存在状态,我们构建了溶剂模型(PCM),溶剂为水(water),文中的图像借助Gaussian-view、Chemoffice2008、Multiwfn2.5等程序[21]进行分析和绘制。
2 结果与讨论
2.1 几何结构分析
为了更好地认识分子的内部结构,如图2所示,将分子的关键原子进行编号,同时进行几何构型的优化计算。
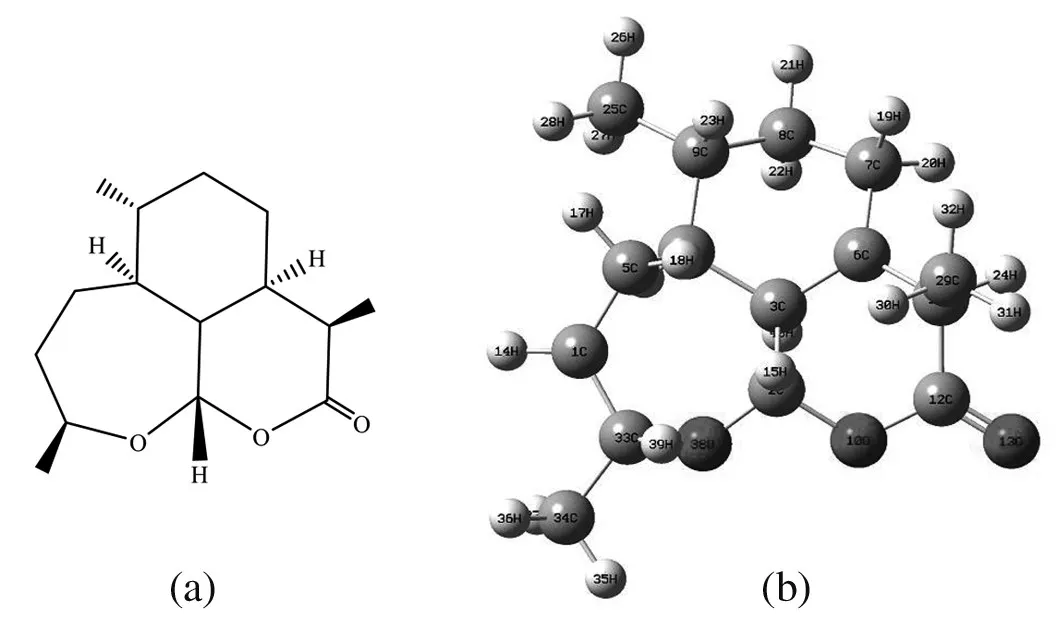
图1 脱氧青蒿素分子的平面结构和立体结构
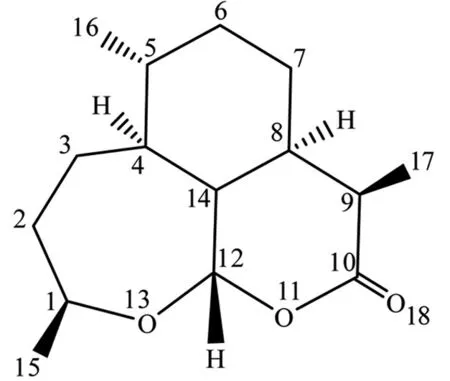
图2 脱氧青蒿素分子的关键原子编号
通过计算,脱氧青蒿素分子中的关键原子之间的键长数据、部分键角数据如表1所示,发现各个类型的键的键长数据与文献[22]上列述的基本一致,七元环(由1,2,3,14,12,13等原子构成)已经严重歪曲而不在一个平面,且C-C单键拉伸处于不稳定状态,六元环(由4,5,6,7,8,14等原子构成)处于较稳定的状态,六元环(由8,9,10,11,12,14等原子构成)也发生了一定的歪曲,虽然键长数据比其他环内的键长数据小,但所有原子并未在同一个平面内。二面角4C-14C-12C-11C的角度为172.4°,二面角8C-14C-12C-13O的角度为-175.2°,二面角4C-14C-8C-9C的角度为147.8°,这些均表明三个环状结构并未形成良好的共轭体系,由于其中大部分碳原子均是单键的饱和结构,所以此分子的共轭效应很弱。从空间构型来看,七元环和酯基很可能是药物分子的活性中心,而上面的六元碳环发挥了一个稳定分子构型的作用。在优化几何构型基础上,继续使用Freq方法进行振动频率分析,其结果全部为正值,表明几何优化的构型是稳定可信的。
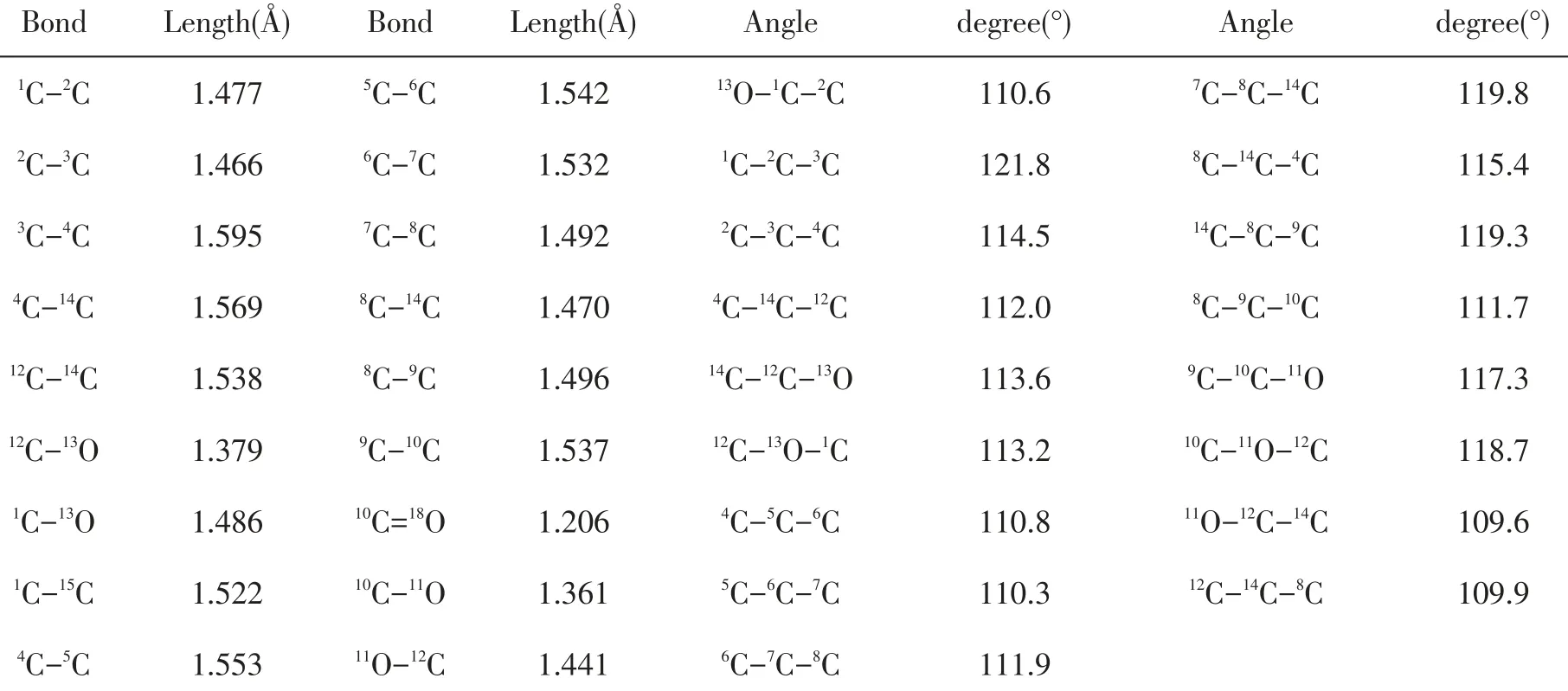
表1 脱氧青蒿素分子中部分键长及键角数据
2.2 紫外吸收光谱
采用TDDFT/B3LYP/6-311+G(d,p)方法结合PCM溶剂模型,基于几何优化结构合理的构型上进行了紫外吸收光谱的理论计算模拟,其结果如图3所示。脱氧青蒿素分子在259.33nm处有强的紫外吸收峰,这是因为酯基的强吸收峰在多环共轭作用下发生了红移;338.40nm处出现较强的紫外吸收峰是由于醚氧键及多环共轭效应的结果;在波长为786.79nm处的可见光区域也显示出吸收峰,这归属于电子从最高占据轨道(HOMO)跃迁到最低最低空轨道(LUMO),这与其分子标准UV-Vis图谱[23]显示的紫外吸收峰吻合较好。
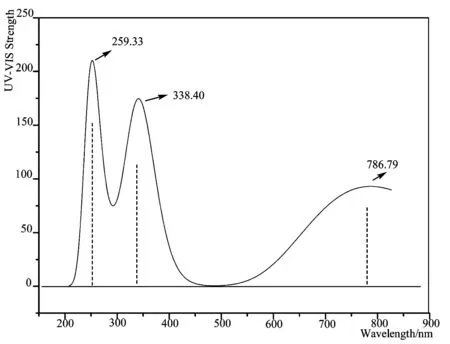
图3 模拟脱氧青蒿素分子的紫外-可见光吸收图
2.3 电子圆二色吸收光谱
采用量子化学密度泛函理论在B3LYP/6-311+G(d,p)水平上对脱氧青蒿素分子的已优化构型进行电子圆二色模拟,如图4所示,脱氧青蒿素分子的ECD谱,252nm处存在着正性康登效应,这是由于分子内还有环状分子轨道上的电子跃迁导致的。同时,我们通过量化计算还预测到目标化合物在212nm处存在负性康登效应,高于400nm整个分子的圆二色性几乎为0,表明官能团的电子跃迁引起的圆二色性效应很弱。
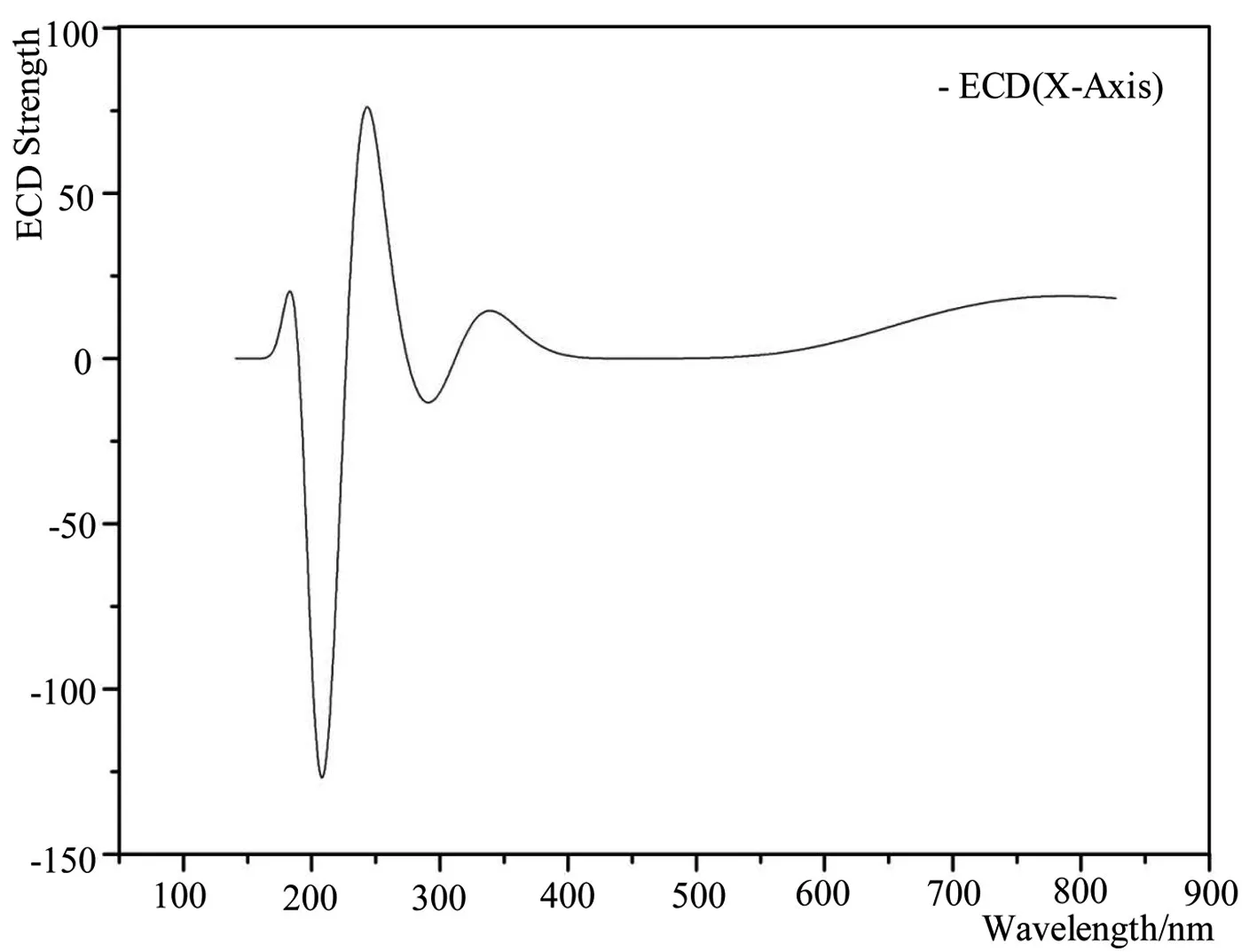
图4 模拟脱氧青蒿素分子的电子圆二色吸收光谱图
2.4 红外吸收光谱
物质的红外吸收光谱是其分子骨架结构的反映,图谱中大多数吸收峰可与各个基团的简谐振动形式对应。我们采用密度泛函理论方法DFT/B3LYP/6-311+G(d,p)结合高斯View程序找出红外光谱中的各个极大点,并描绘成峰如图5所示。
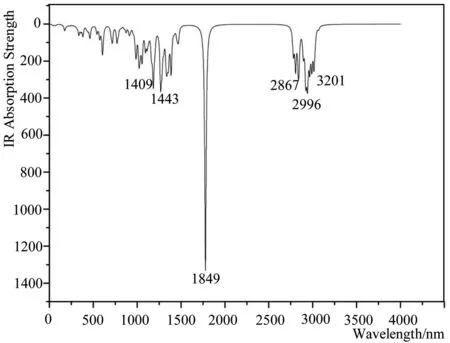
图5 模拟脱氧青蒿素分子的红外光谱图
很显然,脱氧青蒿素分子的红外峰最显著出现在1849nm处,这是由于酯基中C=O双键的伸缩运动导致;2867、2996、3201nm处为分子中甲基的伸缩振动特征峰;1409nm等处出现多个碎峰是由于多元环出现摇摆振动,其中1443nm处为七元环的摇摆振动特征峰,主要体现在3C原子上,这可能是由于空间位阻效应导致的,低于1000nm的峰为C-H键的振动峰,这些峰与实验IR图谱的数值[23,24]基本吻合。
2.5 拉曼吸收光谱
拉曼光谱的分析可以获得更多分子的振动与转动,采用密度泛函理论DFT/B3LYP方法,在6-311+G(d,p)基组水平上进行了分子构型优化和频率分析,得到了多个特征峰。通过计算和比较分别将它们的振动模式进行了详细的归属指认,如图6所示。由于脱氧青蒿素分子中主要存在着酯基,所以在3000cm-1处出现了较多的强峰;分子中有多处CH3和CH2等形成的对称和反对称谱线集中在3300cm-1处;环状结构比较多;各个多元环上的C-H键伸缩振动体现的谱线也集中在3200cm-1处;由于内环变形和扭曲,相关基团的变形振动导致倍频和合频等形式出现,导致1100cm-1-1800cm-1出现连续的碎峰,对比实验拉曼光谱和理论拉曼光谱,它们的吸收峰情况是基本吻合的[23,24]。
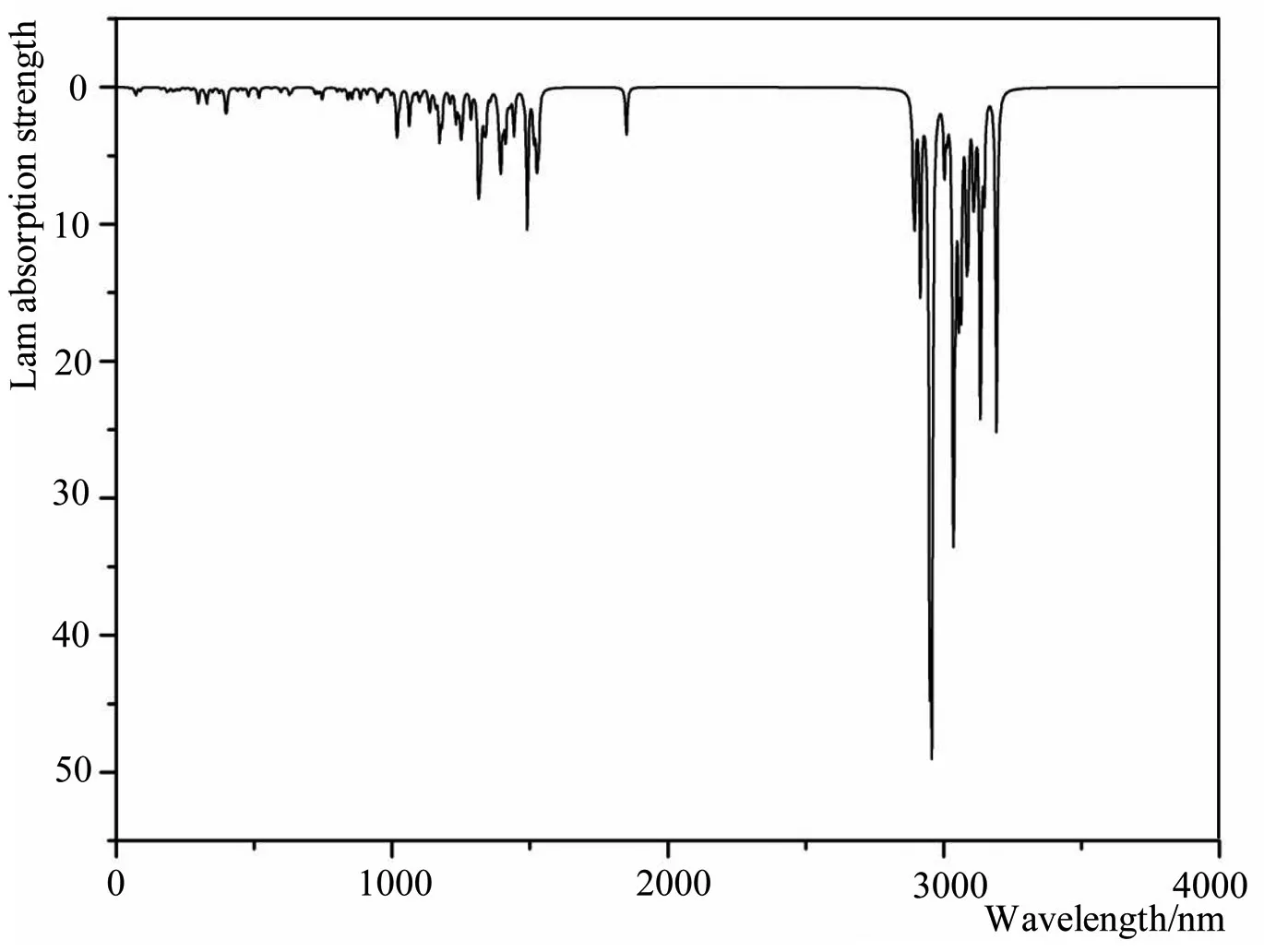
图6 模拟脱氧青蒿素分子的拉曼光谱图
2.6 核磁共振光谱
脱氧青蒿素分子的氢核共振谱(1HNMR,如图7所示),甲基上3个氢(弱,2.32-2.62×10-6);环上CH2中的氢(中,3.38-3.84×10-6);多环交叉共用的H(中,5.85-6.58×10-6);受O原子的强电子吸引作用,旁边的氢(中,5.42-8.95×10-6)。分子中各个链上的C—H上的H原子的化学位移发生了类似的变化,这与受强吸电子基的影响和多环共用碳原子有密切的联系,其化学位移数值基本符合规律,这些进一步说明了我们的模拟计算的可行性。
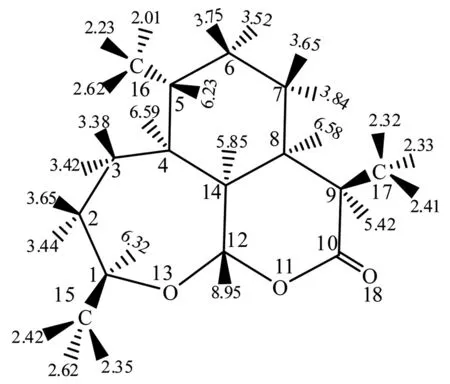
图7 模拟脱氧青蒿素分子的核磁共振氢谱
2.7 荧光发射光谱
脱氧青蒿素分子中含有多个环,且具有酯基、醚氧基等结构,这些导致它可以制备良好的光学性质材料。用CIS方法找到脱氧青蒿素分子的第1激发态,然后使用TDDFT/B3LYP/6-311+G(d,p)的方法结合PCM模型,对它的分子荧光进行了理论模拟计算。
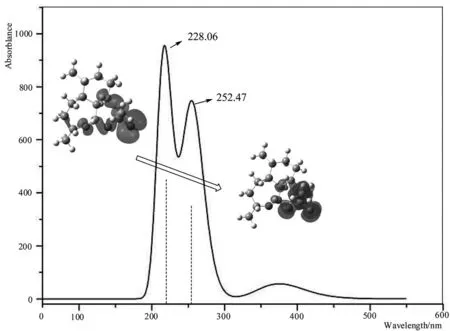
图8 模拟脱氧青蒿素分子的荧光发射光谱
图8描绘的是直接TDDFT方法直接寻找的第1激发态所产生的荧光发射光谱图,很明显在228.06nm、252.47nm处出现了共振荧光光谱峰,分析其主要做贡献的是脱氧青蒿素分子中酯基的反键→成键轨道上,这说明官能团效应占主要地位。已知第69轨道是HOMO轨道,而70是LUMO轨道。将228.06nm、252.47nm的峰中,做主要贡献的分子轨道图如表2中数据所示。其中,酯基结构的反键轨道上电子激发后发生荧光发射在荧光发射中发挥了主体地位,其主要波长出现约在228.06nm处;分子轨道跃迁70→69是LUMO→HOMO轨道上的发射跃迁,在贡献中做了主要作用;分子轨道跃迁70→68是LUMO→HOMO-1轨道上的发射跃迁,体现了电子经荧光发射落到了分子的三环共轭轨道上;分子轨道跃迁70→68是LUMO→HOMO-2轨道上的发射跃迁,体现了电子经荧光发射落到了分子的两环共轭轨道上,其中七元环中的O原子轨道占较大比例。量子化学模拟计算发现,脱氧青蒿素分子的发光主要归属于酯基上的π-π*跃迁所为,但七元环中C-O-C键也能参与荧光发射,这些光谱较紫外吸收光谱进行了一定程度的蓝移,符合了大多数有机分子激发态电子荧光发射跃迁的基本特征[25]。

表2 脱氧青蒿素分子的荧光发射计算分析
2.8 振动圆二色吸收光谱
振动圆二色吸收光谱的测定在分子内基团的旋光和折光率方面有准确而宽广的用途,我们使用与之前计算相同的方法和基组计算了脱氧青蒿素分子的振动圆二色吸收光谱。如图9所示,(a)是指0-4000nm段的脱氧青蒿素分子的振动圆二色吸收光谱图,(b)是指200-1600nm段的它的振动圆二色吸收光谱图。很明显,脱氧青蒿素分子的酯基、醚氧基作为紫外生色基团,吸收光后会产生光偏转导致出峰;脱氧青蒿素分子的多环互相靠近重叠,导致有一定的螺旋式结构且残基的种类和数目均大,故在振动圆二色谱峰中出现较多的碎杂峰,其峰主要集中1000-1600nm段。
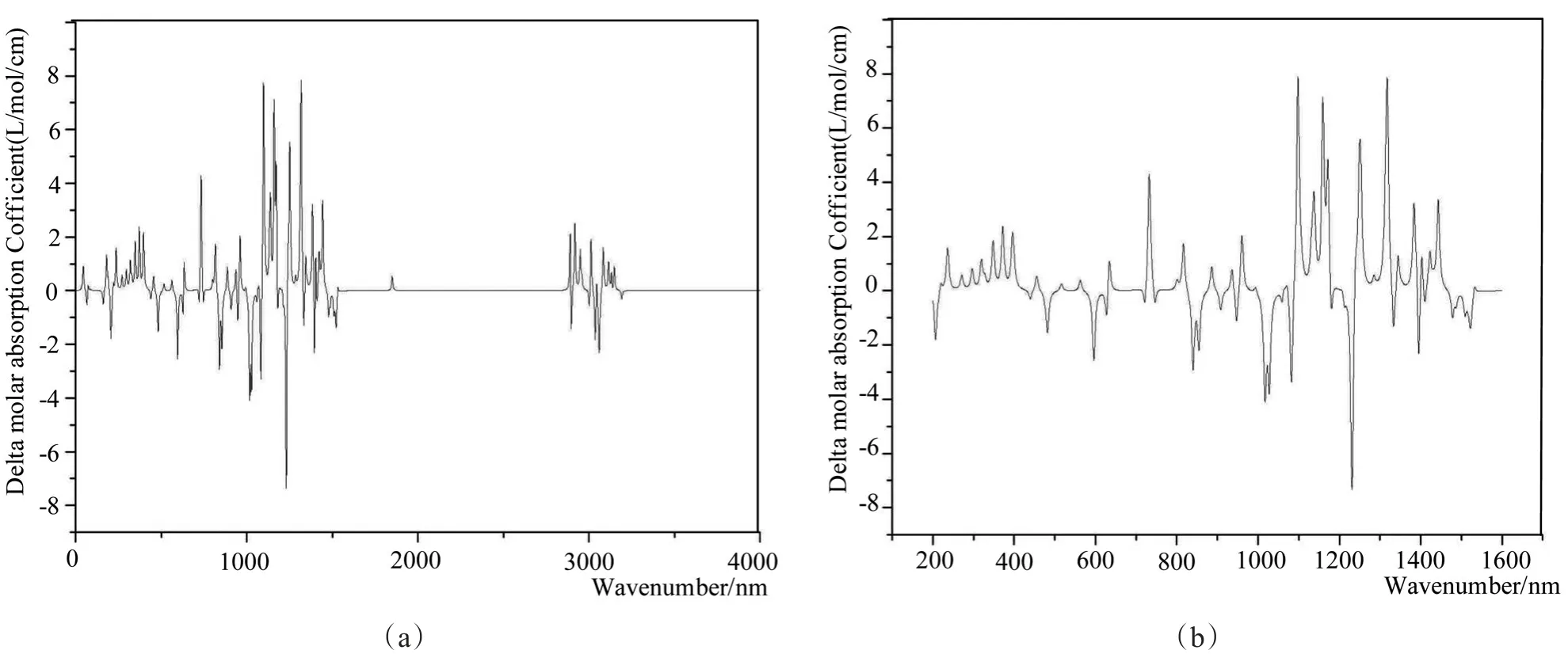
图9 模拟脱氧青蒿素分子的振动圆二色吸收光谱图
2.9 反应活性
由于分子的结构较为复杂且分子体积较大,单从分子轨道方面无法全面地认识分子的化学活性高低,尝试发现原子的自然电荷数值,以进一步揭露分子的活性区域,特别是亲核与亲电特性强弱。在相同的计算方法和基组水平上,将分子中部分原子的自然电荷数值列于表3,其中13O原子所带的电荷最负(-0.490e),其次是11O原子所带的电荷(-0.463e),预示着这2个原子可能是亲电试剂进攻的部位;其中10C原子所带的电荷最正(0.605e),其次是12C原子所带的电荷(0.384e),显然前者是由于所连O原子的较大电负性所导致,由于12C原子是两环共用,所受的空间位阻效应的影响也比较大,此两处原子较难作为亲电试剂去进攻其他分子。Fukui函数图10也表明,2C、8C等原子的成键轨道为蓝色区域,表明此处是亲电试剂最易进攻的部位;2C、8C等原子的反键轨道为绿色区域,表明此处是亲核试剂最易进攻的部位;而其他原子发挥亲电、亲核的反应可能性非常小。f(2)所示的函数图表明,脱氧青蒿素分子的药理活性的发挥与2C、8C等原子关联程度最大,其中2C原子的反键轨道为绿色,表明该部位为亲核试剂的主要活性位点,而8C原子的反键轨道为蓝色,表明该部位为亲电试剂的主要活性位点。虽然与NBO分析中原子的电荷值表明的意义不一致,但这更能对认识药物分子的活性提供科学指导依据。
表3 脱氧青蒿素分子的部分自然原子电荷值
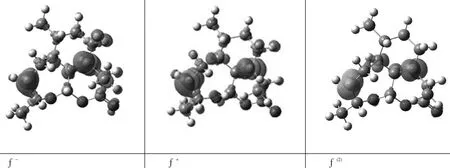
图10 脱氧青蒿素分子的Fukin函数图
3 结论
以脱氧青蒿素分子为主要研究对象,采用密度泛函理论和方法,在6-311+G(d,p)基组水平上进行了优化计算。通过对其分子光谱(UV-Vis谱,ECD谱,IR,拉曼光谱,1HNM,VCD谱以及荧光光谱等)进行了理论模拟和指认,取得了与实验数据基本吻合的结果。通过自然原子电荷(NBO)计算表明,11O、13O等原子很可能是亲电试剂的活性位点,整个分子很难作为亲电试剂去进攻其他分子。Fukui函数图揭露了2C、8C原子在脱氧青蒿素分子中发挥药效的活性中心,使得脱氧青蒿素分子的特殊结构能协同发挥广泛药效奠定基础。文章采用理论模拟光谱峰及其指认将为脱氧青蒿素分子的物证分析与检测、分子结构与性质用途提供有益的理论指导意义。
[1]Chang ZY.ThediscoveryofQinghaosu(artemisinin)asan effectiveanti-malariadrug:auniqueChinastory[J].SciChina Life Sci,2016,59:81-88.
[2]董贻诚,赫荣乔.通力合作攻难关—青蒿素立体结构解析回眸有感[J].生物化学与生物物理进展,2015,42(10):969.
[3]屠呦呦.青蒿及青蒿素类药物[M].北京:化学工业出版社,2009:1-4.
[4]Winzeler EA.Malaria research in the post-genomic era[J].Nature,2008,(455):751-756.
[5]TheraM A,Plowe CV.Vaccines forMalaria:How closearewe[J].Annu RevMed,2012,(63):345-357.
[6]Mott BT,Tripathi A,Siegler MA,etal.Synthesis and antimalarial efficacy of two-carbon-linked,artemisinin-derived trioxane dimers in combinationwith known antimalarialdrugs[J].JMed Chem,2013,56(6):2630-2641.
[7]郭宗儒,青蒿素类抗疟药的研制[J].药学学报,2016,51(1):157-164.
[8]何林,傅川,曾明辉.青蒿素及其衍生物的抗肿瘤作用研究进展[J].华西药学杂志,2015,30(4):517-520.
[9]HoWe,Peh Hy,Chan Tk,etal.Artemisinins:pharmacologicalactionsbeyond anti-malarial[J].Pharmacol Ther,2014,142(1):126-139.
[10]汪大婷,吴明,等.青蒿素模拟热量限制延长酵母寿命的双期响应模式全转录组谱解析[J].中国科学:生命科学,2015,45(4):398-412.
[11]盛庆寿,王武,等.双氢青蒿素对原发性肝癌大鼠的治疗作用及机制[J].中国实验方剂学杂志,2014,20(14):150-154.
[12]邵义如,朱尤庆,等.二氢青蒿素对结肠癌细胞系SW 480增殖凋亡的影响[J],武汉大学学报,2008,29(3):319-323.
[13]王花丽,宋纪蓉,等.甲酰基硫脲类嘧啶衍生物结构与活性的理论研究[J].计算机与应用化学,2005,22(7):531-536.
[14]LU Zhencheng,PENGYonghong.A theoretical elucidation on radicalscavenging activity of three anthocyanins in black rice[J].Natural Product Research and Development,2005,17(6):688-690.
[15]陈莹,徐抗震,等.酚酸抗氧化活性的理论计算[J].食品科学,2011,32,(9):36-39.
[16]龙威,几种常见镇痛药分子活性的DFT及DRFT研究[J].首都师范大学学报(自然科学版),2015,36(5):46-52.
[17]龙威,王榆元,谭倪,等.过渡金属Co、Mo促进噻吩裂解反应的密度泛函研究[J].新疆师范大学学报(自然科学版),2015,34(2):30-35.
[18]Frisch M J,TrucksGW,SchlegelH B,etal.Gaussian03,Revision D.02;Gaussian,Inc.:Pittsburgh,PA,2003.
[19]ParrRC,PariserR.StructureTheory[J].ConceptsandMethodsinModern TheoreticalChemistry:Electronic Structureand Reactivity,2013:431.
[20]LiY,Evans JN S.The Fukui function:A key concept linking frontiermolecular orbital theory and the hard-soft-acidbase principle[J].Journalof the American ChemicalSociety,1995,117(29):7756-7759.
[21]Lu T,Chen F.Multiwfn:Amultifunctionalwavefunction analyzer[J].Journalof Compu tationalChemistry,2012,33(5):580-592.
[22]J.A.Dean.Lange’sHandbook ofChemistry[M].McGraw-HillBook Company.1985:913-1032.
[23]SpectralDatabase forOrganic CompoundsSDBS[DB/OL].http://sdbs.db.aist.go.jp/.
[24]Hu Shufang,Feng Yanyan,Zhang Daosen,etal.Raman spectral changesof Artemisinin-induced Rajicellsapoptosis[J].VibrationalSpectroscopy,2015:81,83-89.
[25]陆维敏,谱学基础与结构分析[M].北京:高等教育出版社,2005.
Density Functional Analysis and Spectral Properties Identification of Artemisinin
LONGWei,YANG Xing-yue,JIANG Luo-ying,CHEN Yu-yu,SHANG Xin-yu
(University ofSouth China,Chemical Engineering,Hengyang,Hunan,421001,China)
The density functional theory DFT/B3LYP/6-311+G(d,p)method and basis setwere used to carry out theoretical simulation and identification for UV-Vis spectroscopy,ECD spectroscopy,IR spectroscopy,Raman spectroscopy,1HNMR spectrum and Fluorescence spectroscopy of artemisinin.2C and 8C atomsmay be the electrophilic and nucleophilic reaction center,which can play pharmacological activity by Fukui function scanning.The synergistic effect is also existed between themanymultiple ring.All that can play amore effective pharmacological activity,all these can become a scientific and reasonable theoreticalmodelwhich can perform the pharmacological activity bymodern computationalchemistry.
Artemisinin;Density functional theory;Electronic spectra;Quantum chemistry
O641,TQ 25
A
1008-9659(2016)02-0001-09
2016-03-20
衡阳市科技局项目(2015KJ09)。
龙 威(1983-),男,湖南湘潭人,博士,实验师,主要从事催化材料、药物活性的量化研究。
猜你喜欢
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中国资源综合利用(2017年1期)2018-01-22
山东工业技术(2016年15期)2016-12-01
华人时刊(2016年1期)2016-04-05
中国粮油学报(2016年5期)2016-01-23
——青蒿素
中国学术期刊文摘(2015年21期)2015-12-24
中国品牌(2015年11期)2015-12-01
郑州大学学报(理学版)(2014年3期)2014-03-01
