
煤焦油加氢脱氧精制研究进展
2016-09-02 06:07许人军胡薇月崔文岗李稳宏
广州化工 2016年15期
许人军,胡薇月,崔文岗,李 冬,李稳宏
(1 西安市轻工业研究所,陕西 西安 710001;2 陕西延长石油油田化学科技有限责任公司,陕西 延安 716000;3 陕西省产品质量监督检验研究院,陕西 西安 710048;4 西北大学化工学院,陕西 西安 710069)
煤焦油加氢脱氧精制研究进展
许人军1,2,胡薇月3,崔文岗4,李冬4,李稳宏4
(1 西安市轻工业研究所,陕西西安710001;2 陕西延长石油油田化学科技有限责任公司,陕西延安716000;3 陕西省产品质量监督检验研究院,陕西西安710048;4 西北大学化工学院,陕西西安710069)
简要介绍了煤焦油中主要含氧化合物的类型,包括酚类、萘类、酯类、酸类、呋喃类和醛类等含氧化合物,综述了不同类型含氧化合物在加氢脱氧(HDO)过程中的反应机理以及反应路径,总结了加氢脱氧过程中直接脱氧和间接脱氧的相互关系以及路径选择,最后针对煤焦油中含氧化合物复杂及多样性的特点提出了一些新的改进和研究方向,并对含氧化合物加氢脱氧的未来作了展望。
加氢脱氧;含氧化合物;反应机理;动力学
随着世界石油资源的匿乏以及能源需求的不断增加,面对我国“富煤,少气,缺油”的能源现状,近年来,人们已经开始寻求和开发新的可替代能源。煤液化油、煤焦油等因其产量不断增加,越来越引起人们的注意,其可谓理想的能源替代品,然而这些新型可替代能源已存在着诸多加工问题,如高的杂原子含量,相对于原油来说,煤焦油中氧含量极高,这使得在生产清洁燃料油品之前必须将氧脱除[1-4]。因此,对煤焦油进行加氢脱氧研究具有重要的经济和战略意义。
本文对主要油品中含氧化合物类型、加氢脱氧反应机理以及加氢脱氧反应动力学的研究进展进行了综述,并对未来的研究方向进行了展望。
1 含氧化合物的类型
原料中氧含量和氧化物的类型决定了实现较高的加氢脱氧(HDO)转化率时的氢耗和操作难度[5]。在轻馏分加氢中,HDO并不是很重要,但在重质油加氢催化改质过程中很重要。
HDO是煤液化油生产燃料产品中最重要的反应之一。液化方法和煤的结构决定了氧化物的类型。为了研究其加氢过程中的HDO反应,Gates等[6-8]对由溶剂精炼煤法(SRC)生成的煤液化油进行了大量表征。这些学者使用制备液相层析法从SRC液体里分出了九个馏分段,5,6,7,8-四氢化-1-萘酚,2-羟苯基苯,4-环己基苯基苯酚等酚类化合物主要集中在弱酸馏分段中。其他的含氧化合物,如呋喃类,醚类和酮类集中在中性油馏分段中,在碱性馏分段发现了羟基吡啶和羟基吲哚。
耿层层等[9]对低温煤焦油中的含氧化合物进行了分析鉴定。此外,吴婷等[10]采用GC-MS及元素分析仪对低温煤焦油中酸性组分和碱性组分的化学组成和结构进行定性定量分析。其中,酸性组分质量分数不小于0.1%以上的化合物有74种,且全部为含氧化合物,其质量分数为95.4%。
2 加氢脱氧的反应机理
2.1呋喃类
Furimsky[11-13]使用Co-Mo/Al2O3催化剂对呋喃的HDO反应进行了研究,提出了呋喃HDO反应机理,如图1所示。在上述反应机理中,可利用的表面活化氢是影响反应途径的决定性因素。途径1为在一定的反应条件下氧从环中脱出;途径2为在活化氢浓度更高的情况下,在发生开环反应之前呋喃环产生了部分加氢反应,在后续的反应中生成丙烯或生成丁烯;途径3为在较高氢分压下的反应,在这种条件下催化剂表面大部分被活化氢所覆盖,呋喃环可以完全加氢,主要反应产物丁烷和H2O。
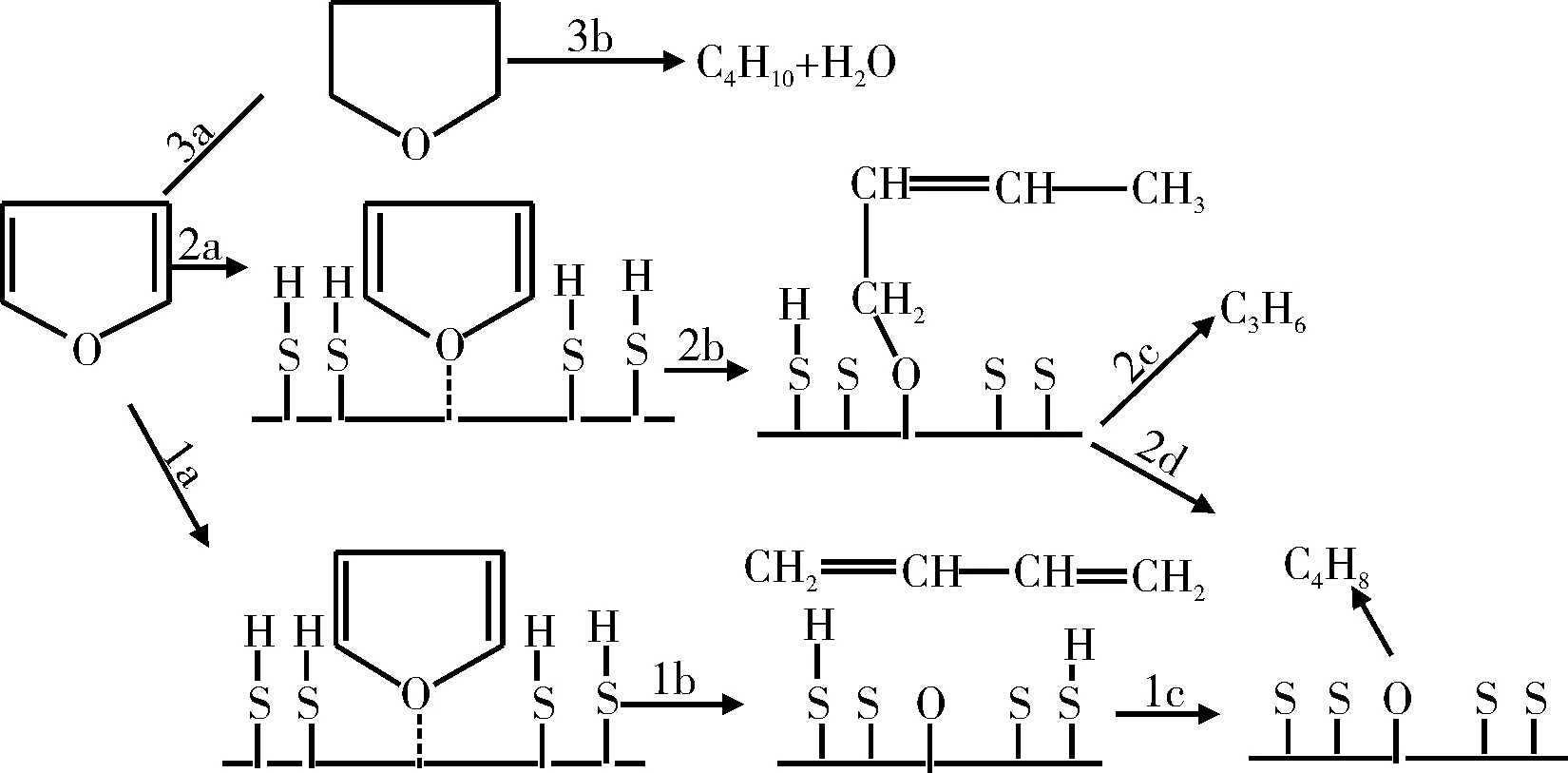
图1 呋喃HDO反应途径Fig.1 HDO reaction pathway of furan
Bunch等[14-15]使用硫化态和还原态Ni-Mo/Al2O3催化剂研究了苯并呋喃的HDO反应,提出的反应机理示于图2。研究结果表明,使用硫化态Ni-Mo/Al2O3催化剂,生成约50%的乙基苯酚及少量的乙苯和乙基环己烷,并且只发现一种加氢含氧中间产物2,3-二氢苯并呋喃。然而,使用还原态Ni-Mo/Al2O3催化剂,苯并呋喃加氢脱氧反应只有氢化反应,经HDO反应生成含氧中间产物的类型较多(如六氢苯并呋喃、2-乙基环己醇和八氢苯并呋喃),中间产物再脱氧生成乙基环己烯和乙基环己烷,产物中并没有发现乙基苯酚和乙苯。
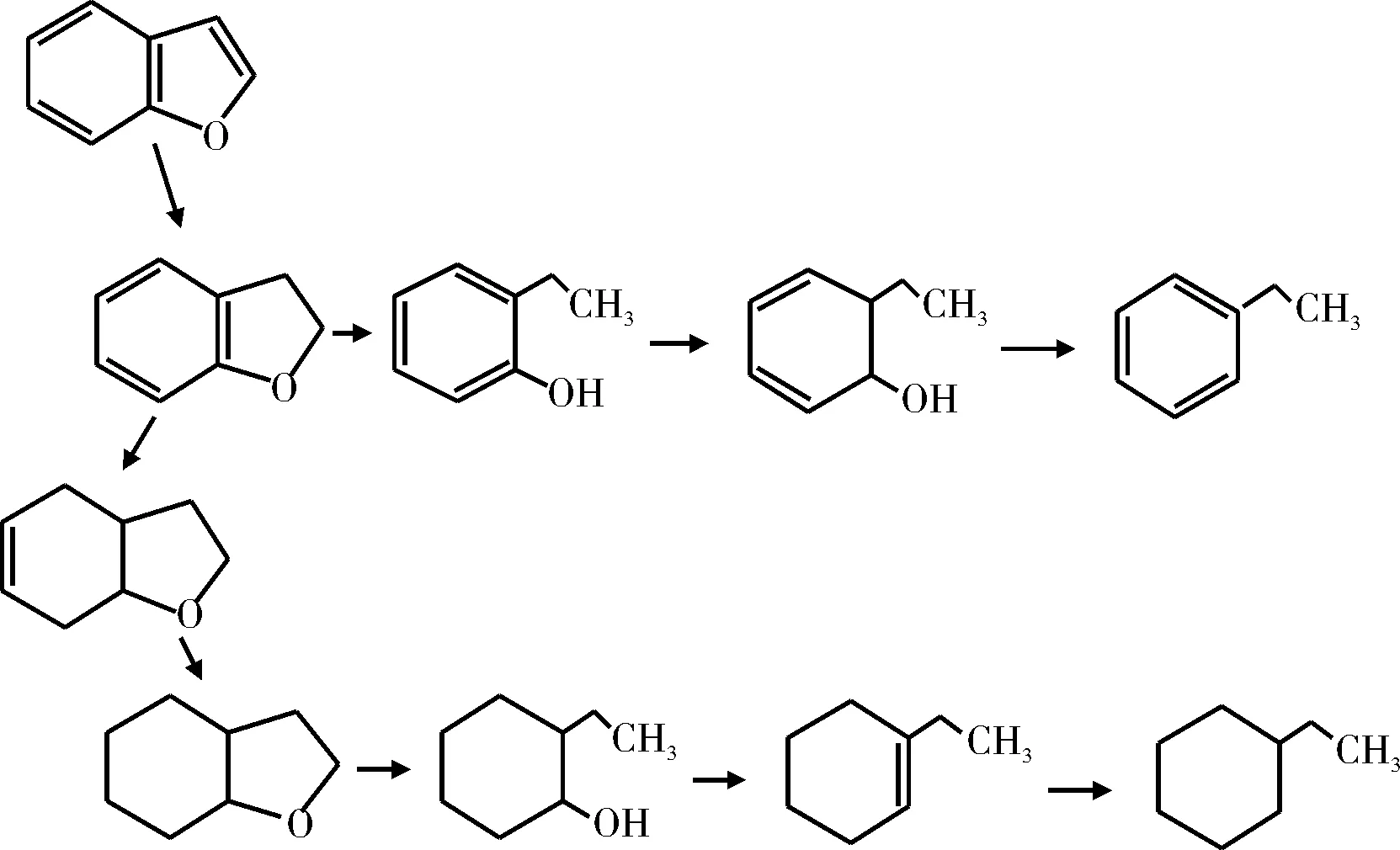
图2 苯并呋喃的HDO反应网络Fig.2 HDO reaction network of benzofuran
Krishnamurthy等[16]根据二苯并呋喃HDO的产物分布和某些中间体的反应活性,提出了二苯并呋喃的HDO反应机理,见图3。二苯并呋喃的HDO主要按照3种途径进行:(1)加氢途径:首先芳环加氢饱和生成含氧中间产物,然后C-O键断裂生成单环烃;(2)氢解途径:先发生C-O键断裂生成苯基苯酚,再加氢生成单环产物;(3)直接脱氧途径:二苯并呋喃直接脱氧生成联苯。在较高的氢分压下,在杂环开环以前,首先进行的是一个芳环的加氢反应。在反应中生成了邻-环乙基苯酚中间产物,其进一步脱氧生成二环己烷或是苯基环己烷。这就说明,与相应的含硫化合物HDS反应相比,HDO反应需要更高的氢耗[17]。
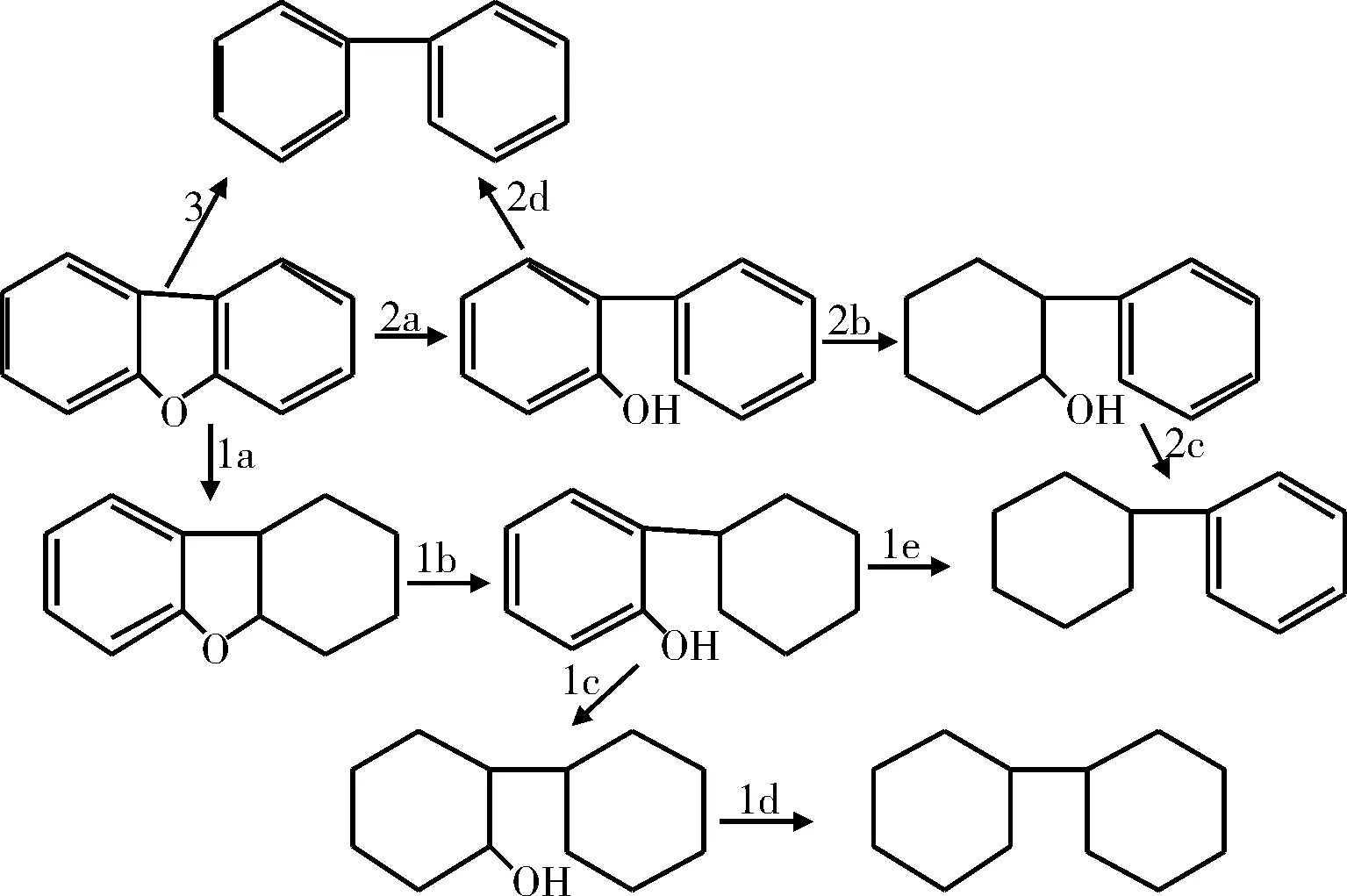
图3 二苯并呋喃HDO反应网络Fig.3 HDO reaction network of diphenylene-oxide
Satterfield等[18]对苯并呋喃的HDO进行了研究,发现反应条件对产物分布的影响非常显著。Lavopa等[19]研究了硫化态和氧化态Ni-Mo/Al2O3催化剂对苯并呋喃的加氢脱氧反应。使用硫化态催化剂催化反应,产物中单环烃类占75%,环己烷占主要部分,还有一些重要但含量少的物质如甲基环戊烷、环戊烷、苯、甲基环己烷和环己烯等。然而,使用氧化态催化剂催化反应,单环化合物收率为25%。
2.2酚类化合物
Odebunmi等[20-22]在连续微型反应器内,使用硫化态Co-Mo/Al2O3催化剂在氢分压为3.0~12.0 MPa下,研究了甲基苯酚的HDO反应途径,研究发现主要加氢产物是甲苯和环己烷,同时生成了少量环己烯。Samchenko等[23]使用Ni/Cr催化剂研究发现邻甲基苯酚和对甲基苯酚比间甲基苯酚更稳定。Shin等[24]使用Ni/SiO2催化剂建立了下面的加氢反应顺序:苯酚≈间甲基苯酚>对甲基苯酚>邻甲基苯酚。研究结果均表明,邻位取代酚的位阻效应影响很大[25]。
Wandas等[26]使用Co-Mo/Al2O3催化剂,在氢气压力为7 MPa,温度为360 ℃条件下研究了甲酚的HDO反应。同样,直接脱氧生成了许多种类的化合物,且甲基环己烷、甲苯和乙基环戊烷是主要的加氢脱氧产物。Furimsky等[27]采用硫化态Co-Mo/Al2O3催化剂,对邻位和对位取代苯酚的HDO反应进行了研究。研究结果表明,邻位取代苯酚的HDO反应活性最弱,苯酚、对乙基苯酚和对叔丁基苯酚HDO反应的转化率基本相同。邻位取代苯酚的HDO反应主要按两种途径进行如图4和图5所示。
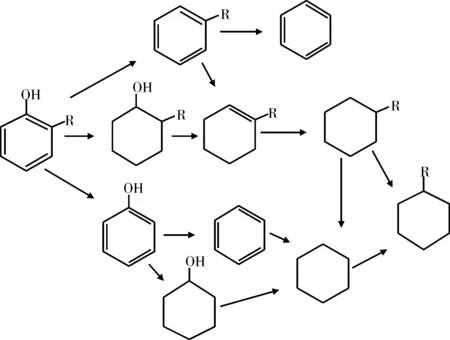
图4 邻位取代苯酚的HDO反应网络Fig.4 HDO reaction network of ortho substituted phenol
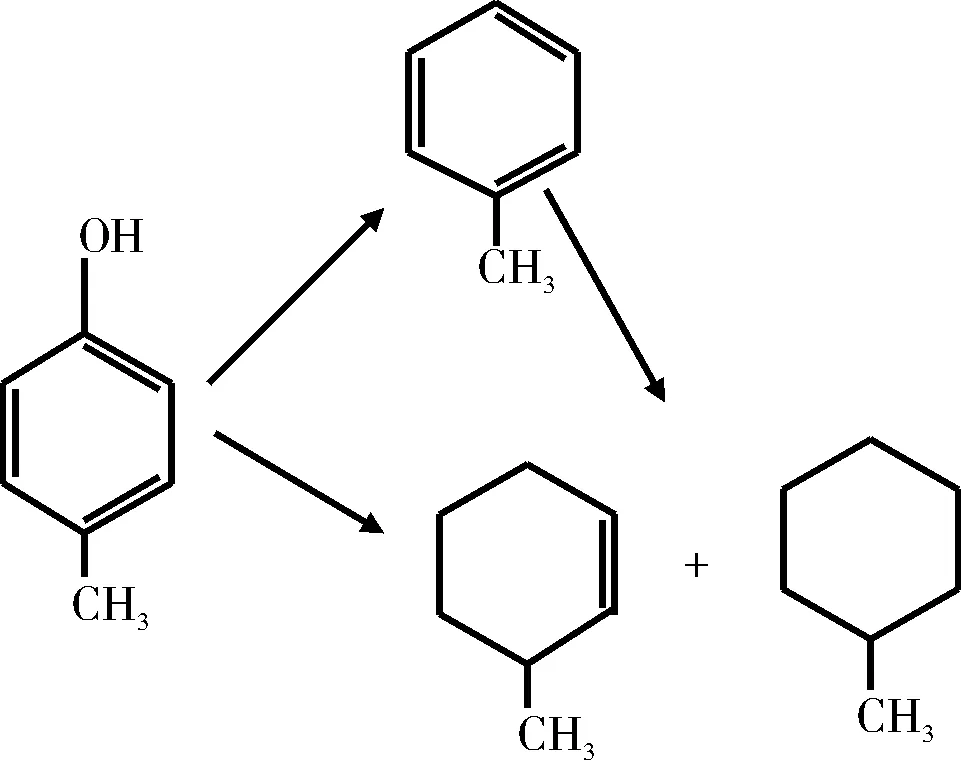
图5 对甲基苯酚的HDO反应网络Fig.5 HDO reaction network of p-methylphenol
Vogelzang等[28]提出的萘酚的HDO反应网络(图6),反应网络中四氢萘酮生成速率最大,表明该反应的最终产物大部分由四氢萘酮转化而来。该试验使用硫化态Ni-Mo/Al2O3作为催化剂,反应温度为200 ℃,氢气压力为3.5 MPa。在此条件下,芳香环的加氢饱和比萘酚的直接HDO更容易进行。因此,四氢化萘和5,6,7,8-四氢-1-萘酚占萘酚HDO产物很大一部分比例。然而,在高温下条件下萘酚的直接HDO速率超过了芳环的加氢饱和。同时,他提出包含1,2-二氢萘酚和四氢萘酮、酮-烯醇的转化是反应网络的一部分。而且,顺式和反式十氢化萘的生成速率非常低。
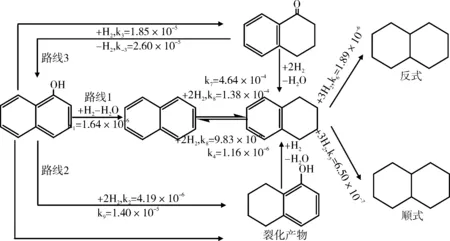
图6 萘酚的HDO反应网络Fig.6 HDO reaction network of naphthol
徐春华[29]研究了不同反应温度和压力下邻甲酚在硫化态Co-Mo/Al2O3催化剂上的HDO性能,反应中直接脱氧产物甲苯的选择性高达90%。Lin等[30-31]研究了Rh基催化剂上邻甲氧基苯酚的HDO反应过程。研究结果表明,邻甲氧基苯酚在Rh基催化剂上是按先加氢后脱氧的途径进行反应,反应过程见图7。王威燕等[32]研究了270 ℃下非晶态催化剂La-Ni-Mo-B催化4-甲基苯酚的HDO反应(见图8)。在整个反应过程中,没有检测到甲苯的生成,这说明在La-Ni-Mo-B催化下,4-甲基苯酚没有发生直接脱氧反应,而是先加氢生成环己醇,再脱氧生成甲基环己烷,即按照氢化-氢解的途径进行反应。
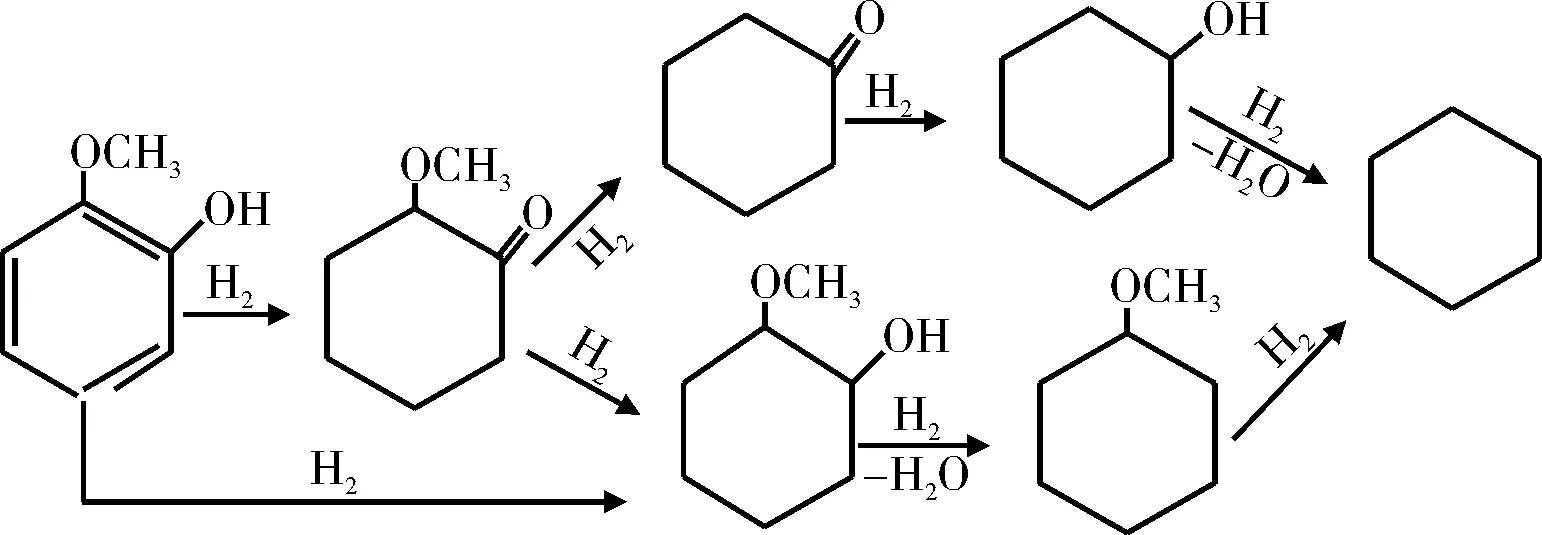
图7 邻甲氧基苯酚在Rh基催化剂上的HDO反应途径Fig.7 HDO reaction pathway on Rh based catalysts of o-methoxyphenol
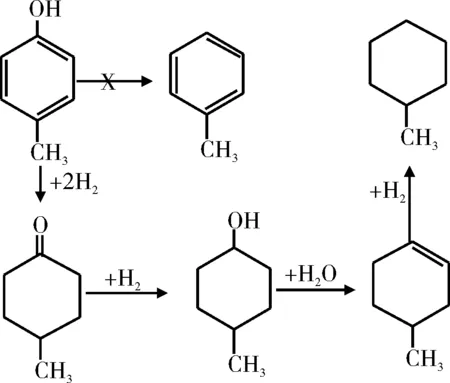
图8 非晶态催化剂La-Ni-Mo-B催化4-甲基苯酚的HDO反应路径Fig.8 Reaction scheme of HDO of 4-methyl phenol on La-Ni-Mo-B amorphous catalysts
Bredenberg等[33]在氢压5 MPa和温度(275~325)℃条件下,研究硫化态Co-Mo/γ-Al2O3催化剂催化3种甲氧基苯酚异构体的加氢脱氧反应,3种异构体的反应活性顺序为对甲氧基苯酚>邻甲氧基苯酚>间甲氧基苯酚。Kallury等[34]研究了Mo-Ni/Al2O3催化剂催化二羟基苯异构体的加氢脱氧反应,结果表明,间苯二酚活性较弱,邻苯二酚和对苯二酚的活性比苯酚强,催化脱除一个羟基得到苯酚的收率为60%。
2.3醚类化合物
醚在原料中的含量相对较少,此外,在含羟基的加氢脱氧反应中也可能生成醚。Artok等[35]在温度范围为375~425 ℃,氢气压力为6.9 MPa,含有MoS2的条件下研究了二苯醚的HDO过程。二苯醚的醚键首先加氢裂化生成了苯和酚,并通过将酚转化苯和环己烷以及环己烷的异构化反应生成甲基环戊烷完成整个反应。类似的,Petrocelli和Klein[36]使用硫化态Co-Mo/Al2O3催化剂,在压力为7.0 MPa下发现酚和苯是二苯醚HDO反应的主要产物。在高的转化率下(温度高于300 ℃),酚被转化为苯和环己烷(见图9),这与Shabtai等人[37]的结果一致。
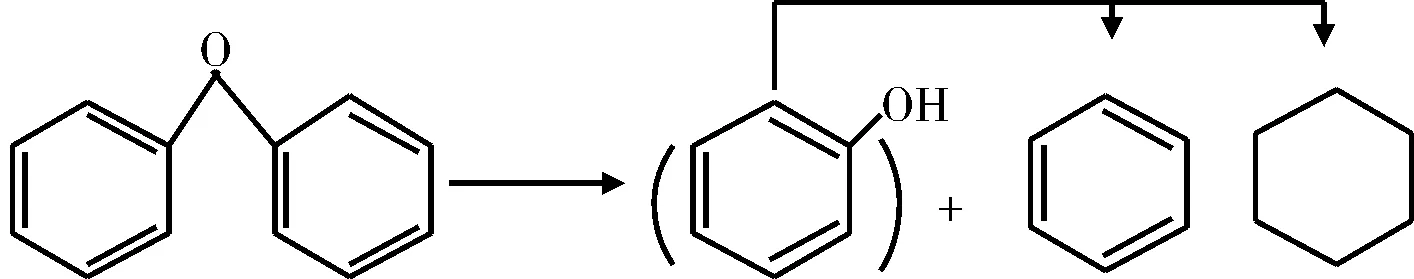
图9 二苯醚的HDO反应机理Fig.9 HDO reaction mechanism of two phenyl ether
2.4酮类
酮类主要有两种HDO反应途径:(1)直接氢解生成烃类化合物;(2)先加氢生成醇,再氢解生成烃类化合物(见图10)。
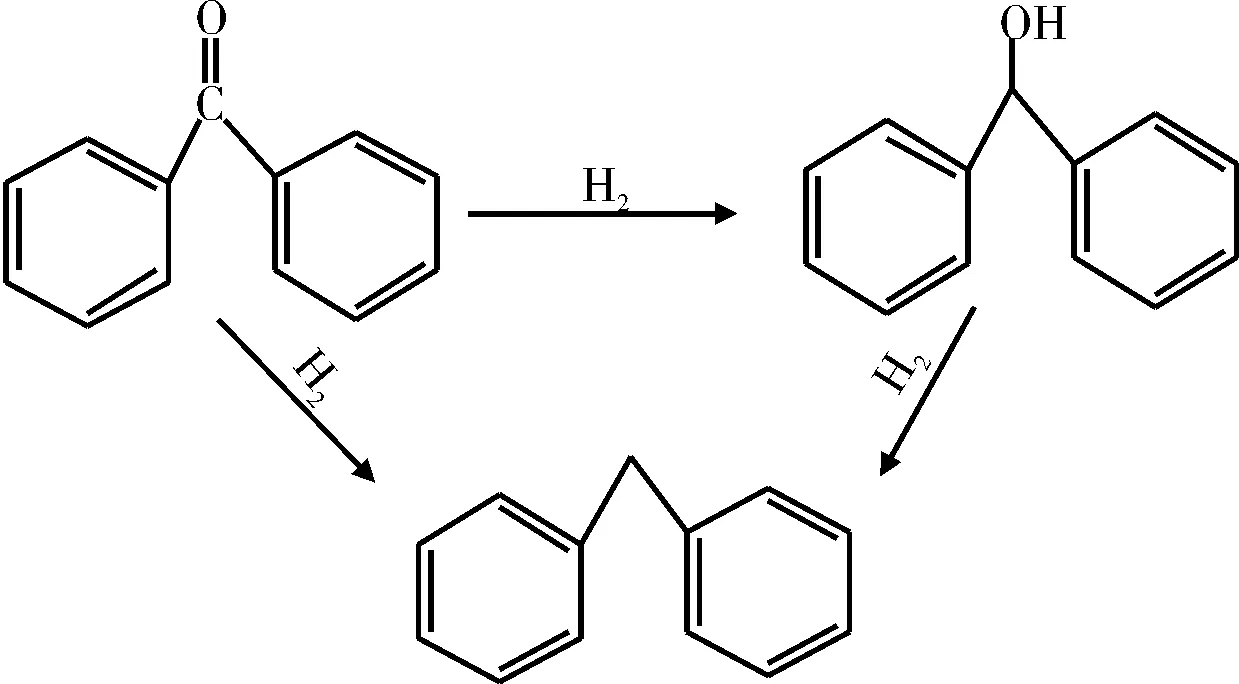
图10 二苯甲酮的HDO反应途径Fig.10 HDO reaction pathway of benzophenone
Durand等[38]在250 ℃下用Ni-Mo/γ-Al2O3催化剂催化环己酮加氢脱氧反应,环己烷为主要的反应产物,反应收率达95%。Oliva等[39]认为,在金属中心催化环己酮C=O氢化生成环己醇,然后在酸中心脱水生成环己烯。不同载体对酮的加氢脱氧反应影响较大。Puente等[40]用硝酸对活性炭进行了改性处理,结果表明,在280 ℃,催化反应120 min,对甲基苯乙酮转化率达100%,对甲基乙基苯为主要产物。
3 结语与展望
近年来,众多研究者对模型含氧化合物加氢脱氧的机理等做了大量研究。但在含氧化合物的HDO反应过程中,由于采用的催化剂类型不同,反应途径和产物的选择性也有所不同。但是到目前为止,并未指出采用不同类型的催化剂所得产物选择性有较大差异的本质原因。以后可从以下三个方面进行研究和改进:(1)进一步详细研究含氧化合物在不同类型催化剂上的HDO反应机理,找到导致反应产物选择性不同的本质原因;(2)开发新型的选择性更好的适合煤焦油及模型化合物加氢脱氧催化剂;(3)通过大量含氧模型化合物HDO反应的研究,探究清楚实际油品中不同类型含氧化合物在HDO过程中的相互作用,为实际油品的HDO反应和工业化生产提供依据。
[1]Furimsky E. Catalytic hydrodeoxygenation[J]. Applied Catalysis A: General, 2000, 199(2): 147-190.
[2]Robert J A. An overview of modeling studies in HDS,HDN and HDO catalysis[J]. Polyhedron,1997, 16(18): 3073-3088.
[3]Ellott D C, Neuenschwander G G. Liquid fuels by low severity hydrotreating of biocrude[J]. Developments in Thermochemical Biomass Conversion, 1996, 1(2): 611-621.
[4]Grange P, Laurent E, Maggi R, et al. Hydrotreatment of pyrolysis oils from biomass: reactivity of the various categories of oxygenated compounds and preliminary techno-economical study [J]. Catal Today, 1996, 29(4): 297-301.
[5]Furimsky E. Chemistry of catalytic hydrodeoxygenation [J]. Catalysis Reviews: Science and Engineering, 1983, 25(3): 421-458.
[6]Grandy D W, Petrakis L, Young D C. Determination of oxygen functionalities in synthetic fuels by NMR of naturally abundant 17O[J]. Nature Y, 1984, 308(5955): 175-177.
[7]Petrakis L, Young D C, Ruberto R G. Catalytic hydroprocessing of SRC-Ⅱ heavy distillate fractions. 2.Detailed structural characterizations of the fractions[J]. Industrial and Engineering Chemistry Process Design and Development Y, 1983, 22(2): 298-305.
[8]Petrakis L, Ruberto R G, Young D C, et al. Catalytic hydroprocessing of SRC-Ⅱ heavy distillate fractions. 1.Preparation of the fractions by liquid chromatography[J]. Industrial and Engineering Chemistry Process Design and Development Y, 1983, 22(2): 292-298.
[9]耿层层,李术元,岳长涛,等.神木低温煤焦油中含氧化合物的分析与鉴定[J].石油学报,2013,29(1):130-136.
[10]吴婷,凌凤香,马波,等.GC-MS分析低温煤焦油酸性组分及碱性组分[J].石油化工离等学校学报,2013,26(3):44-52.
[11]Furimsky E. Mechanism of catalytic hydrodeoxygenation of tetrahydrofuran[J]. Industrial and Engineering Chemistry Product Research and Development A, 1983, 22(1): 31-34.
[12]Furimsky E. Deactivation of molybdate catalyst during hydrodeoxygenation of tetrahydrofuran[J]. Industrial and Engineering Chemistry Product Research and Development A, 1983, 22(1): 34-38.
[13]Furimsky E. The mechanism of catalytic hydrodeoxygenation of furan[J]. Applied Catalysis A, 1983, 6(2): 159-164.
[14]Bunch A Y, Ozkan U S. Investigation of the reaction network of benzofuran hydrodeoxygenation over sulfided and reduced Ni-Mo/Al2O3catalysts[J]. Journal of Ctalysis(Print) A, 2002, 206(2): 177-187.
[15]Bunch A Y, Wang X Q, Ozkan U S. Hydrodeoxygenation of benzofuran over sulfided and reduced Ni-Mo/γ-Al2O3catalysts: effect of H2S[J]. Journal of Molecular Catalysis A, Chemical A, 2007,270 (1-2): 264-272.
[16]Krishnamurthy S, Panvelker S, Shah Y T. Hydrodeoxygenation of dibenzofuran and related compounds[J]. AIChE Journal A, 1981, 27(6): 994-1001.
[17]Badilla O R, Pratt K C, Trimm D L. A study of nickel-molybdate coal-hydrogenation catalysts using model feedstocks[J]. Fuel A, 1979, 58(4): 309-314.
[18]Satterfield C N, Yang S H. Simultaneous hydrodenitrogenation and hydrodeoxygenation of model compounds in a trickle bed reactor[J]. Journal of Catalysis A, 1983, 81(2): 335-346.
[19]Lavopa V, Satterfield C N. Catalytic hydrodeoxygenation of dibenzofuran[J]. Energy and fuels A, 1987, 1(4): 323-331.
[20]Odebunmi E O, Ollis D F. Catalytic hydrodeoxygenation.1.Conversions of o-,p-,and m-cresols[J]. Journal of Catalysis, 1983, 80(1): 56-64.
[21]Echeandia S, Arias P L, Barrio V L, et al. Synergy effect in the HDO of phenol over Ni-W catalysts supported on active carbon:effect of tungsten precursors[J]. Applied Catalysis B, Environmental A, 2010, 101(1-2): 1-12.
[22]Gevert S B, Eriksson M, Eriksson P. Direct hydrodeoxygenation and hydrogenation of 2,6-and 3,5-dimethylphenol over sulphided CoMo catalyst[J]. Applied Catalysis A: General, 1994, 117(2): 151-162.
[23]Samchenko N P, Pavlenko N V. Reactivity of alkylphenols in liquid phase catalytic hydrogenation[J]. Reaction Kinetics and Catalysis Letters A, 1982, 18(1-2): 155-158.
[24]Shin E J, Keane M A, Catalytic hydrogen treatment of aromatic alcohols[J]. Journal of Catalysis (Print) A, 1998, 173(2): 450-459.
[25]Weigold H. Behaviour of Co-Mo-Al2O3catalysts in the hydrodeoxygenation of phenols[J].Fuel A,1982,61(10):1021-1026.
[26]Wandas R, Surygala J, Sliwka E. Conversion of cresols and naphthalene in the hydroprocessing of three-component model mixtures simulating fast pyrolysis tars[J]. Fuel (Guildford) A, 1996, 75(6): 687-694.
[27]Furimsky E. Catalytic hydrodeoxygenation[J]. Applied Catalysis A: General A, 2000, 199(2): 147-190.
[28]Vogelzang M W, Li C L, Schuit G C A, et al. Hydrodeoxygenation of 1-naphthol: activities and stabilities of molybdena and related catalysts[J]. Journal of catalysis (Print) A, 1983, 84(1): 170-177.
[29]徐春华.加氢脱氧反应对硫化态催化剂结构的影响[D].北京:石油化工科学研究院,2011.
[30]Senol O I, Ryymin E M, Viljava T R. Effect of hydrogen sulphide on the hydrodeoxygenation of aromatic and aliphatic oxygenates on sulphided catalysts[J]. Journal of Molecular Catalysis. A, Chemical A, 2007, 277(1-2): 107-112.
[31]Lin Y C, Li J L, Wan H P. Catalytic Hydrodeoxygenation of Guaiacol on Rh-Based and Sulfided CoMo and NiMo Catalysts[J]. Energy Fuels, 2011, 25(3): 890-896.
[32]王威燕,杨运泉,罗和安,等.La-Ni-Mo-B非晶态催化剂的制备加氢脱氧性能及失活研究[J].燃烧化学学报,2011,39(5):367-372.
[33]Bredenberg J B S, Huuska M, Toropainen P. Hydrogenolysis of differently substituted methoxyphenols[J]. Journal of Catalysis (Print) A. 1989, 120(2): 401-408.
[34]Kallury R K M R, Wanda M. Hydrodeoxygenation of hydroxy, methoxy, and methyl phenols with molybdenum oxide/nickel oxide/alumina catalyst[J]. Journal of Catalysis (Print) A, 1985, 96(2): 535-543.
[35]Artok L, Erbatur O, Schobert H H. Reaction of dinaphthyl and diphenyl ethers at liquefaction conditions[J]. Fuel processing technology A, 1996, 47(2): 153-176.
[36]Petrocelli F P, Klein M T. Modeling lignin liquefaction. I: Catalytic hydroprocessing of lignin-related methoxyphenols and interaromatic unit linkages[J]. Fuel Science and Technology International A, 1987, 5(1): 25-62.
[37]Shabtai J, Nag N K, Massoth F E. Catalytic functionalities of supported sulfides. IV: C-O hydrogenolysis selectivity as a function of promoter type[J]. Journal of Catalysis (Print) A, 1987, 104(2): 413-423.
[38]Durand R, Geneste P, Moreau C. Hetergeneous hydrodeoxygenation of ketones and alcohols on sulfided NiO-MoO3/γ-Al2O3catalyst[J]. Journal of Catalysis, 1984, 90(1): 147-149.
[39]Olivas A, Samano E C, Fuentes S. Hydrogenation of cyclohexanone on nickel-tungsten sulfide catalysts[J]. Applied Catalysis A:General, 2001, 220(1-2): 279-285.
[40]De L P, Gil G, Pis A J. Effects of support surface chemistry in hydrodeoxyg-enation reactions over CoMo/activated carbon sulfided catalysts[J]. Langmuir, 1999, 15(18): 5800-5806.
Research Progress on Carl Tar Hydrodeoxygenation
XURen-jun1,2,HUWei-yue3,CUIWen-gang4,LIDong4,LIWen-hong4
(1 Xi’an Institute of Light Industry,Shaanxi Xi’an 710001;2 Shaanxi Extend Oil Oilfield Chemical Technology Co., Ltd., Shaanxi Xi’an 716000; 3 Shaanxi Province Supervision and Inspection Institute of Product Quality,Shaanxi Xi’an 710048;4 School of Chemical Engineering, Northwest University,Shaanxi Xi’an 710069,China)
The types of the main oxygen-containing compounds in coal tar were briefly described, including phenols, naphthalene, esters, acids, aldehydes, furans and other oxygen-containing compounds, and the reaction mechanisms about different types of oxygen-containing compounds in hydrodeoxygenation(HDO) process was simply introduced. Meanwhile, reaction mechanism and pathway of different types of oxygen-containing compounds were also reviewed. The directly and indirectly in the process of HDO and path selection were summarized. For a complexity and diversity of oxygenated compounds contained in the coal tar, a number of improvements and new research directions were made. In addition, the prospect of oxygen-containing compounds HDO was discussed.
hydrodeoxygenation(HDO);oxygenated compound;reaction mechanism;kinetics
李稳宏(1955-),男,博导。
TQ517.4
A
1001-9677(2016)015-0039-05
猜你喜欢
中学化学(2022年5期)2022-06-17
科学导报(2022年28期)2022-05-24
能源化工(2021年6期)2021-12-30
化工管理(2021年7期)2021-05-13
云南化工(2020年11期)2021-01-14
合成技术及应用(2021年1期)2021-01-07
环境保护与循环经济(2020年4期)2020-06-08
中国特种设备安全(2019年1期)2019-03-13
中学生数理化·高二版(2016年3期)2016-12-26
中学生数理化·高二版(2016年3期)2016-12-26
