
一种芽孢杆菌中性蛋白酶启动子的克隆和验证
2016-09-13 01:20纪明华赵利超史吉平孙俊松
食品工业科技 2016年13期
纪明华,赵利超,史吉平,孙俊松,*
(1.中国科学院上海高等研究院,上海 201210;2.中国科学院大学,北京 100049;3.上海大学,上海 200444)
一种芽孢杆菌中性蛋白酶启动子的克隆和验证
纪明华1,2,赵利超1,3,史吉平1,孙俊松1,*
(1.中国科学院上海高等研究院,上海 201210;2.中国科学院大学,北京 100049;3.上海大学,上海 200444)
为研究1398中性蛋白酶高效表达的分子机制,对其生产菌株进行了基因组测序和比对,确认了表达该中性蛋白酶的操纵子序列及结构。通过构建1398中性蛋白酶表达质粒,将其转入枯草芽孢杆菌;以gfp为报告基因,将包含P1398在内的不同启动子分别构建到质粒pMK4,转入枯草芽孢杆菌。发酵研究结果表明:P1398是一种底物自发诱导的高效启动子,是菌株表达1398中性蛋白酶的关键因素。1398中性蛋白酶在枯草芽孢杆菌中可获得稳定的表达量,其中在164中活性最高(1210 U/mL);重组枯草芽孢杆菌在LB中培养时,P1398介导的GFP的表达量低于P43和PxylA,而在工业培养基PPB中,其表达量为PxylA介导的GFP的2.14倍。因此,P1398可用于食品工业酶重组生产菌株的构建。
解淀粉芽孢杆菌,中性蛋白酶,启动子,gfp
解淀粉芽孢杆菌(Bacillusamyloliquefaciens)是一类与枯草芽孢杆菌(B.subtilis)近缘的芽孢杆菌[1],关于解淀粉芽孢杆菌的研究主要集中在菌种分离纯化及代谢产物特性分析,尤其是抑菌性能方面的研究[2-5]。虽然已经公布了一些解淀粉芽孢杆菌的基因组测序数据[1,6-8],但是对其蛋白表达的分子机制的研究报道不多。
解淀粉芽孢杆菌被认定为安全(Generally Recognized As Safe,GRAS)的微生物,由于外源DNA的转化效率过低,其分子改造十分困难,因此该类菌株目前难以用作重组生产菌株;而枯草芽孢杆菌具有遗传操作工具丰富、遗传背景清晰、蛋白分泌量高等优点,是许多外源分泌蛋白高水平表达的首选宿主细胞[9]。在外源蛋白的重组表达构建时,选择转录水平高的启动子是重要因素之一;已经应用于枯草芽孢杆菌中的成熟启动子主要有P43、PamyQ、Pspac、PxylA等,这些启动子在实验室的富集培养基中表现良好,但鲜见它们被用于酶制剂蛋白的放大生产,它们还存在需使用诱导物、高培养成本或表达效率不高等缺点。
本研究前期研究发现,我国广泛用于生产中性蛋白酶的1398芽孢杆菌[10],其中性蛋白酶产量极高,因此对其开展了分子机理的研究。通过分子生物学手段分析发现1398蛋白酶生产菌株为解淀粉芽孢杆菌,经全基因组测序及序列比对后确认了表达中性蛋白酶(nprE)的操纵子(operon)结构及序列,并通过克隆表达验证了该操纵子的功能。为进一步了解1398 NprE启动子P1398的转录特性,以gfp为报告基因,并以启动子PxylA和P43为对照,在不同培养基中进行了重组菌株发酵的研究。

表1 菌株和质粒

表2 引物列表
1 材料与方法
1.1材料与仪器
菌株与质粒见表1,芽孢杆菌菌株包括解淀粉芽孢杆菌1398和枯草芽孢杆菌1A976(SCK6)、168和164(ATCC 6051a)。其中,1398芽孢杆菌来自白银赛诺生物科技有限公司,164购自美国ATCC菌种保藏中心,168及1A976[11]由BGSC赠予。
基因组DNA提取试剂盒、PCR清洁试剂盒、质粒小剂量提取试剂盒等杭州爱思进生物技术有限公司;DNA聚合酶Prime STAR、DNAligase等TaKaRa公司;限制性内切酶Pst I、EcoR I、BamH I、Xma I等及其体系Thermo Scientific;木糖、干酪素等国药集团化学试剂有限公司。
Synergy H1型酶标仪美国BioTek公司;DU-730型分光光度计美国Beckman Coulter公司。
1.2实验方法
1.2.1引物设计与PCR所扩增DNA片段、对应模板及引物见表2。PCR所用DNA聚合酶为Prime STAR,PCR基本程序如下:预变性98 ℃ 2 min;变性98 ℃ 10 s;退火55 ℃ 10 s;延伸72 ℃,1 kb/min。
1.2.2培养基的配制菌体活化、培养、转化所用培养基为LB培养基,100 mL LB组成为1 g NaCl、1 g胰蛋白胨、0.5 g酵母浸取物;发酵用蛋白酶生产培养基(Protease producing broth,PPB),100 mL蛋白酶生产培养基组成为6.5 g玉米粉、4 g豆粕、5.5 g麸皮、0.3 g Na2HPO4·12H2O、0.03 g KH2PO4;枯草芽孢杆菌1A976诱导转化培养基为含1%(w/v)木糖的LB培养基。用于大肠杆菌转化子的筛选的氨苄青霉素(Amp)浓度为100 μg/mL;用于枯草芽孢杆菌转化子筛选的氯霉素(Cm)浓度为10 μg/mL。
1.2.31398芽孢杆菌基因组的提取、测序及表达质粒的构建接种1398芽孢杆菌单菌落于3 mL LB培养液中,37 ℃,200 r/min培养10~12 h,离心10000×g,1 min,收集菌体,以AxyGEN基因组DNA提取试剂盒及其方法提取基因组DNA。1398芽孢杆菌的基因组测序服务由生工生物工程(上海)股份有限公司提供。芽孢杆菌表达质粒的构建均基于pMK4,目标产物由PCR扩增获得,再经限制性内切酶酶切及T4 DNA连接酶连接后转入大肠杆菌DH5α,质粒提取后由上海生工生物工程有限公司测序验证。
1.2.4感受态的制备及转化大肠杆菌的转化采用氯化钙法[12]。164和168采用电击转化方法[13];1A976在参照原方法[11]的基础上有少量改变,具体方法如下:单菌落1A976接种到3 mL LB试管中,37 ℃、200 r/min培养10~12 h;取300 μL转接到预热的3 mL LB试管中,加入过滤除菌的的木糖溶液至木糖终浓度为1%,37 ℃、200 r/min继续培养3 h后分装到2 mL离心管中,然后加入待转化的质粒混匀,37 ℃、200 r/min混合培养1.5 h后涂含有相应抗生素的LB琼脂平板培养。
1.2.5质粒的提取大肠杆菌质粒的提取采用AxyPrep质粒小剂量提取试剂盒中的方法。枯草芽孢杆菌的质粒提取在试剂盒方法的基础上有所改变,即用Buffer S1悬浮细胞后,加入50 mg/mL的溶菌酶20 μL,混匀,把体系置于37 ℃水浴锅内反应30 min,然后加入Buffer S2,其他操作步骤不变。
1.2.6中性蛋白酶酶活测定按照QB/T1803-1993标准进行,1个酶活单位(U)定义为1 min内,水解酪蛋白产生1 μg酪氨酸所需要的酶量。采用紫外分光光度法,以干酪素为底物,用三氯乙酸终止反应,在275 nm处检测吸光度。具体如下:取发酵48 h的发酵液,根据酶活性梯度稀释到一定倍数后,取2 mL酶稀释液在(30±0.2) ℃水浴锅中预热2 min,然后加入10 g/L酪素溶液2.00 mL,摇匀,在(30±0.2) ℃水浴中反应10 min。再加入0.4 mol/L三氯乙酸4.00 mL,摇匀。最后静止10 min后用中性滤纸过滤,取滤液用10 mm比色皿于275 nm处检测吸光度,由OD275值计算酶活。
1.2.7细胞培养与GFP荧光检测枯草芽孢杆菌的摇瓶发酵培养如下:单克隆菌落接种到3 mL LB培养液,30 ℃、200 r/min培养10 h,然后以1%的体积比接种到含有50 mL培养基的500 mL摇瓶中,30 ℃,200 r/min连续培养,每12 h取一次发酵液样品。
细胞发酵液经梯度稀释后用于GFP的荧光检测,200 μL经10倍,100倍稀释后的菌液置于96孔板中,利用酶标仪进行荧光测定:测量时选择荧光检测模式,读数类型选择“endpoint”,设定激发光波长为485 nm,检测光波长为520 nm,设定测量温度为30 ℃。
2 结果与分析
2.11398芽孢杆菌的菌种鉴定及中性蛋白酶操纵子的鉴定
对1398中性蛋白酶高效表达的分子机制进行研究,首先要获取编码该蛋白酶的完整操纵子结构及序列。在以前的研究中已经通过同源基因获得了编码该中性蛋白酶的ORF序列,并通过质谱方法验证了其氨基酸序列[15];但是不能成功扩增表达该蛋白酶的完整操纵子序列,猜测是其启动子序列与已发布的枯草芽孢杆菌基因组序列同源性不高,因此进行了1398芽孢杆菌的全基因组测序。该测序不仅获得了编码1398 NprE的完整操纵子序列,同时也获取了完整的16S rDNA序列,经过比对发现该生产菌株不是先前报道中的枯草芽孢杆菌[16-17],而是解淀粉芽孢杆菌,如图1所示。

图1 以16S rDNA序列鉴定产1398 NprE的芽孢杆菌的种属关系Fig. 1 16S rDNA analysis of 1398 NprE producing bacillus
从图1可以看出,解淀粉芽孢杆菌属与枯草芽孢杆菌属由共同的原始菌株进化而来,相对于研究比较透彻的解淀粉芽孢杆菌DSM7[14],该1398工业生产菌株与枯草芽孢杆菌168的种属关系更为接近。
完整的1398中性蛋白酶的操纵子结构如图2所示。可以看出,1398中性蛋白酶的操纵子由启动子P1398,信号肽Signal peptide,中性蛋白酶的完整nprE基因序列,以及可能的转录终止子序列组成。

图2 1398中性蛋白酶操纵子示意图Fig. 2 Schematic diagram of 1398 nprE operon注:P1398,1398中性蛋白酶启动子;SP,signal peptide,信号肽;ME,mature enzyme,成熟肽;terminator,终止子。
2.21398中性蛋白酶基因的异源表达

图3 1398中性蛋白酶在枯草芽孢杆菌中的表达Fig.3 Expression of 1398 nprE in B.subtilis注:a,pMK4-1398nprE构建流程。1398nprE经Pst I/Nde I双酶切后连接到pMK4相应位点,在大肠杆菌中得到缺失一个A碱基的质粒,以P10/P11引物PCR后转入枯草芽孢杆菌后校正突变;b,枯草芽孢杆菌提取质粒pMK4-1398nprE电泳图。M:DNA Marker,mut:pMK4-1398nprE-Mut,1:所提取1A976的质粒,2:所提取168的质粒,3:所提取164的质粒;c,重组菌株1A976/168/164发酵产1398中性蛋白酶活性。pMK4:空白质粒对照组,pMK4-1398nprE:实验组。
为进一步验证测序所得到的1398中性蛋白酶操纵子的功能,同时也出于解决原始菌株所产酶制品存在的粘度高,不易分离,酸臭味大等问题的技术需求,开展了NprE在枯草芽孢杆菌的重组表达。如图3所示,本研究首先尝试利用酶切克隆的方法构建表达1398 NprE的穿梭质粒pMK4-1398nprE,但是从大肠杆菌始终提取不到含有正确序列的质粒,只得到一些带有阅读框移码突变的质粒pMK4-1398nprE-Mut。有文献报道,中性蛋白酶的表达对于大肠杆菌具有致死效应[18-20],因此猜测可能是由于这一效应导致pMK4-1398nprE无法在大肠杆菌内构建成功。为验证这个猜想,设计了用于修正点突变的引物,以pMK4-1398nprE-Mut质粒DNA为模板,进行移码突变和修正,然后直接将PCR产物转化} 1A976。从1A976转化子中提取质粒并测序验证,最终获得了不含突变的表达质粒pMK4-1398nprE,如图3(a)和图3(b)所示;而将pMK4-1398nprE再次转化DH5α,还是得不到相应的大肠杆菌抗性菌株,从而基本可以证明,1398 NprE操纵子序列可以在大肠杆菌内获得转录和表达,但该表达产物可能由于不能被有效分泌,因此会在宿主大肠杆菌细胞内产生负效应,抑制菌株增殖。
表达质粒pMK4-1398nprE之后又被分别转入B.subtilis164、168,以分别转化了质粒pMK4的三种重组枯草芽孢杆菌菌株为空白对照组。利用蛋白酶工业生产PPB培养基分别进行了摇瓶发酵和重组酶活的测定,实验发现,重组1398 NprE的酶活在48 h左右达到最大值。
图3(c)为6株菌发酵产中性蛋白酶的酶活测定结果,从图3(c)可以看出,pMK4空白对照组相对于实验组中性蛋白酶表达水平很低,携带质粒pMK4-1398nprE的不同枯草芽孢杆菌均能表达中性蛋白酶,但是存在明显的宿主差异性,其中,在164中1398 NprE的酶活水平最高,约为1210 U/mL发酵液,高出1A976重组菌株发酵水平的1倍以上,比168的发酵酶活高出约50%。上述研究结果表明所克隆的1398 nprE operon在枯草芽孢杆菌中具有转录活性。
2.3表达GFP的重组枯草芽孢杆菌的构建
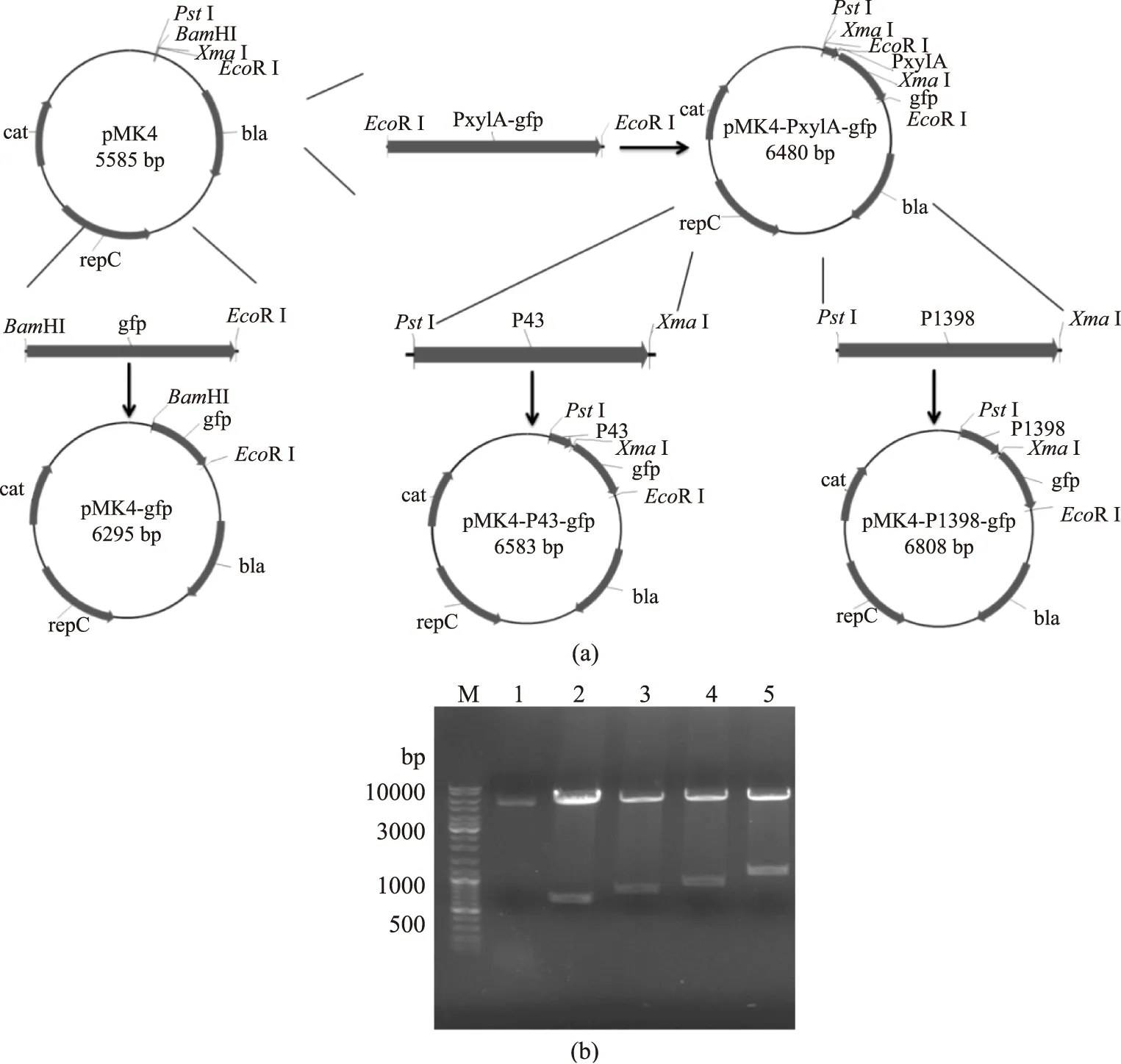
图4 重组质粒的构建Fig. 4 Construction of plasmid注:a,pMK4-gfp、pMK4-PxylA-gfp、pMK4-P1398-gfp、pMK4-P43-gfp构建流程;b,质粒酶切电泳验证。M:DNA Marker,1:pMK4,2:BamH I/EcoR I双酶切pMK4-gfp,3:Pst I/EcoR I双酶切pMK4-PxylA-gfp,4:Pst I/EcoR I双酶切pMK4-P43-gfp,5:Pst I/EcoR I双酶切pMK4-P1398-gfp。
为进一步探究1398中性蛋白酶高表达的分子机制,如图4(a)所示,gfp基因被分别构建到启动子P1398、PxylA以及P43之后,分别得到质粒pMK4-P1398-gfp、pMK4-PxylA-gfp以及pMK4-P43-gfp,pMK4-gfp为不含启gfp基因的动子的对照质粒。pMK4与gfp片段经BamH I、EcoR I双酶切,T4 DNA连接酶连接得到pMK4-gfp;pMK4与DNA片段PxylA-gfp经PstI单酶切,T4 DNA连接酶连接得到pMK4-PxylA-gfp;pMK4-PxylA-gfp与P1398经PstI、XmaI酶切酶连得到pMK4-P1398-gfp;pMK4-PxylA-gfp与P43经PstI、XmaI酶切酶连则得到pMK4-P1398-gfp。质粒构建过程中,除pMK4-gfp外,pMK4-P1398-gfp、pMK4-PxylA-gfp以及pMK4-P43-gfp在大肠杆菌中均能表达GFP,菌落在可见光下呈现绿色,再次验证启动子P1398在大肠杆菌中有转录功能。图4(b)为4个质粒的酶切电泳图,经测序确认质粒序列无误。把pMK4-gfp、pMK4-P1398-gfp、pMK4-PxylA-gfp以及pMK4-P43-gfp分别转化到枯草芽孢杆菌1A976中,即得到4株重组枯草芽孢杆菌。
2.4不同启动子介导GFP表达的发酵培养研究
为了展开对该启动子在枯草芽孢杆菌发酵过程中的表达特性研究,对含有pMK4-gfp、pMK4-P1398-gfp、pMK4-PxylA-gfp以及pMK4-P43-gfp的4株重组菌分别利用实验室常规营养富集培养基LB及蛋白酶工业生产培养基PPB进行摇瓶发酵实验,发酵过程中对GFP的荧光强度进行检测,结果如图5所示。

图5 绿色荧光蛋白在(a)LB和(b)中性蛋白酶生产培养基中的表达Fig. 5 Expression of GFP in(a)Lysogeny broth and(b)protease producing broth注:图中荧光量(Fluorescence intensity)为发酵液稀释100倍后测定结果。
从图5可以看出,在LB和PPB中,三种启动子均能启动gfp的表达。如图5(a)所示,利用LB进行菌株的发酵培养时,P1398启动子引导的GFP表达水平明显不如其它两种启动子,但是在蛋白酶发酵生产的工业培养基PPB中,P1398转录表达的GFP荧光值持续升高(图5(b))。在前24 h内,P1398-GFP的表达量不如PxylA-GFP;而在48 h时,P1398-GFP的表达量比PxylA-GFP高19%,比P43-GFP高48%;待菌株连续发酵到84 h,P1398-GFP的表达量比PxylA-GFP高59%,比P43-GFP高88%;在120 h,P1398-GFP的表达量比PxylA-GFP和P43-GFP分别高出114%和65%。上述结果表明,相对廉价的工业生产培养基PPB对P1398有明显的诱导表达效应。
3 结论
本研究基于16S rDNA比对分析发现1398不是传统认为的枯草芽孢杆菌,而是一种解淀粉芽孢杆菌;从1398解淀粉芽孢杆菌中克隆出的1398nprEoperon(DDBJ/EMBL/GenBank accession no. KU312310),在大肠杆菌中直接表达对宿主具有明显的致死效应;1398解淀粉芽孢杆菌的中性蛋白酶基因可在枯草芽孢杆菌中异源表达,表达量约为1210 U/mL发酵液;本研究首次报道了一种新型启动子P1398,其具有底物自发诱导效应,对菌株1398高产中性蛋白酶有重要作用,以GFP为报告蛋白进行分析时,在粗放培养基中其效率是PxylA的2.14倍。
因此启动子P1398具有良好的应用前景,本研究将继续对该启动子展开突变和优化工作,并将其用于食品加工领域各类酶制剂产品的生产菌株的分子构建研究。
[1]Chen X H,Koumoutsi A,Scholz R,et al. Comparative analysis of the complete genome sequence of the plant growth-promoting bacterium Bacillus amyloliquefaciens FZB42[J]. Nature biotechnology,2007,25(9):1007-1014.
[2]Chowdhury S P,Hartmann A,Gao X W,et al. Biocontrol mechanism by root-associated Bacillus amyloliquefaciens FZB42-a review[J]. Frontiers in microbiology,2015,6:1-11.
[3]王英国,王军华,权春善,等. 解淀粉芽孢杆菌抗菌活性物质的分离纯化及抑菌活性研究[J]. 中国生物工程杂志,2007,27(12):41-45.
[4]张娟,杨彩梅,曹广添,等. 解淀粉芽孢杆菌及其作为益生菌的应用[J]. 动物营养学报,2014,26(4):863-867.
[5]孙力军,陆兆新,孙德坤. Bacillus amyloliquefaciens ES-2 液体发酵抗菌脂肽培养基及其主要影响因子筛选[J]. 食品工业科技,2008(5):60-63.
[6]Gamez R M,Rodríguez F,Bernal J F,et al. Genome sequence
of the banana plant growth-promoting rhizobacterium Bacillus amyloliquefaciens BS006[J]. Genome announcements,2015,3(6):e01391-15.
[7]Rückert C,Blom J,Chen X H,et al. Genome sequence of B. amyloliquefaciens type strain DSM7 T reveals differences to plant-associated B. amyloliquefaciens FZB42[J]. Journal of biotechnology,2011,155(1):78-85.
[8]Cai J,Liu F,Liao X,et al. Complete genome sequence of Bacillus amyloliquefaciens LFB112 isolated from Chinese herbs,a strain of a broad inhibitory spectrum against domestic animal pathogens[J]. Journal of biotechnology,2014,175:63-64.
[9]van Dijl J M,Hecker M. Bacillus subtilis:from soil bacterium to super-secreting cell factory[J]. Microb Cell Fact,2013,12(3):1-6.
[10]胡学智,王俊. 蛋白酶生产和应用的进展[J]. 工业微生物,2008,38(4):49-61.
[11]Zhang X Z,Zhang Y H P. Simple,fast and high-efficiency transformation system for directed evolution of cellulase in Bacillus subtilis[J]. Microbial biotechnology,2011,4(1):98-105.
[12]Sambrook J,Russell David W. 分子克隆实验指南(第三版)[M].北京:科学出版社,2002,96-99.
[13]Xue G P,Johnson J S,Dalrymple B P. High osmolarity improves the electro-transformation efficiency of the gram-positive bacteria Bacillus subtilis and Bacillus licheniformis[J]. Journal of Microbiological Methods,1999,34(3):183-191.
[14]Zhang Y,Li S,Liu L,et al. Acetoin production enhanced by manipulating carbon flux in a newly isolated Bacillus amyloliquefaciens[J]. Bioresource technology,2013,130:256-260.
[15]赵忠润,陈超,黄娇芳,等. 1398 中性蛋白酶的色谱柱纯化与分子鉴定[J]. 生物技术进展,2014,4(3):201-206.
[16]程池,刘光全,李金霞,等.55株芽孢杆菌16S rRNA基因序列测定与系统发育学分析[J]. 食品与发酵工业,2006,32(10):20-24.
[17]陈雪,侯红漫,陈莉,等.产细菌素芽孢杆菌的筛选及发酵条件的研究[J].中国酿造,2008,27(9):26-30.
[18]Wang L F,Ekkel S M,Devenish R J. Expression in Escherichia coli of the Bacillus subtilis neutral protease gene(NPRE)lacking its ribosome binding site[J]. Biochemistry international,1990,22(6):1085-1093.
[19]刘白玲,张义正.枯草杆菌中性蛋白酶基因在大肠杆菌中的表达[J]. 生物工程学报,1997,13(3):304-308.
[20]Wang H,Yang L,Ping Y,et al. Engineering of a Bacillus amyloliquefaciens Strain with High Neutral Protease Producing Capacity and Optimization of Its Fermentation Conditions[J]. PloS one,2016,11(1).
Characterization of a neutral protease promoter derived from aBacillusamyloliquefaciensstrain
JI Ming-hua1,2,ZHAO Li-chao1,3,SHI Ji-ping1,SUN Jun-song1,*
(1.Shanghai Advanced Research Institute,Chinese Academy of Sciences,Shanghai 201210,China;2.University of Chinese Academy of Sciences,Beijing 100049,China;3.Shanghai University,Shanghai 200444,China)
To obtain the DNA elements responsible for transcriptional regulation of the gene expressing 1398 neutral protease(nprE),the genome sequencing of the native bacillus strain was performed and analyzed,the complete operon including upstream region of the possible location of 1398nprEcassette was introduced intoB.subtilisfor heterologous expression.gfpwas then used as the reporter gene for full investigation of the promoter of 1398nprE(P1398)and was compared with known promoter PxylAand P43. It was found that,P1398 was an efficient and substrate-induced promoter as well as a key factor in the expression of 1398 NprE. Stable expression of 1398 NprE inB.subtiliswas detected and the highest enzyme activity was 1210 U/mL in 164. Compared with PxylAand P43,the advantage of P1398 was its capability of bringing better yield of GFP protein inB.subtiliswith medium used for industrial production of enzymes,the GFP level was 214% of that mediated by PxylA,therefore,P1398 was a good candidate of promoter choice for construction of recombinant enzymes’ production using Bacillus.
B.amyloliquefaciens;neutral protease;promoter;gfp
2016-01-14
纪明华(1991-),男,硕士研究生,研究方向:微生物代谢工程,E-mail:15227257110@163.com。
孙俊松(1974-)男,博士,研究员,研究方向:酶工程与生物化工,E-mail:sunjs@sari.ac.cn。
上海市科技攻关计划项目(15295810600)。
TS201.3
A
1002-0306(2016)13-0192-06
10.13386/j.issn1002-0306.2016.13.030
猜你喜欢
湖南饲料(2021年4期)2021-10-13
农药科学与管理(2019年6期)2019-11-23
农药科学与管理(2019年6期)2019-11-23
农药科学与管理(2019年8期)2019-11-23
英语学习(上半月)(2019年9期)2019-10-10
扬子江(2019年3期)2019-05-24
浙江农业学报(2017年1期)2017-05-17
米娜·女性大世界(2016年8期)2016-08-17
工业设计(2016年11期)2016-04-16
当代化工研究(2016年7期)2016-03-20
