
基于Map Reduce的多序列星比对方法在肿瘤研究中的应用△
2016-10-18 01:28李大鹏鞠颖邹权
癌症进展 2016年6期
李大鹏 鞠颖 邹权
1秦皇岛市第四医院肿瘤内科,河北秦皇岛066000
2厦门大学计算机科学系,福建厦门361005
3天津大学计算机科学与技术学院,天津300072
基于Map Reduce的多序列星比对方法在肿瘤研究中的应用△
李大鹏1鞠颖2邹权3#
1秦皇岛市第四医院肿瘤内科,河北秦皇岛066000
2厦门大学计算机科学系,福建厦门361005
3天津大学计算机科学与技术学院,天津300072
序列比对是生物信息学的基础,通过多条序列比对可以挖掘出生物序列中的各种重要信息。大规模的基因序列比对方法对运算能力要求较高,基于Map Reduce框架的多序列比对方法在多序列星比对算法的基础上利用分布式并行计算来处理大规模数据。实验结果表明:相对于单机处理方法,基于Map Reduce的序列比对方法可以更快速地处理大规模数据,并且具有良好的硬件扩展性。本文探讨了多序列比对在肿瘤研究方面的应用前景。
生物信息学;多序列比对;星比对算法;Map Reduce;并行计算;癌症
序列比对是通过对基因序列的比较找出序列之间的相似性和同源性,以便通过已知序列的结构和功能信息来预测未知基因序列所具有的生物学信息。双序列比对通过添加空格或者删除字母使得给定的两条序列具有最大的相似性。多序列比对同时比对多条序列,使得这组序列之间具有最大的相似性。目前已有许多序列比对软件被开发出来,如Clustal[1]、T-coffee[2]、MAFFT[3]和HMMER[4]等。但是随着序列数目的急剧增加,海量的数据对这些软件的计算成本造成了严峻的挑战,因此开发出一种能处理大规模数据并具有良好扩展性的软件是一项重要的工作。
在多序列比对算法方面,目前已取得了一定的成果。基于关键字树的DNA多序列星比对算法[5]利用Aho-Corasick搜索算法(简称AC自动机)进行模式匹配,以便于选取中心序列,进一步用星比对算法进行序列比对。还有研究人员提出利用启发式算法解决多序列比对问题,包括基于A*算法的启发式算法[6],基于模拟退火的多序列比对算法[7],基于遗传算法的多序列比对算法[8],基于粒子群算法的多序列比对算法[9]。也有研究人员利用概率模型进行多序列比对,如基于隐马尔可夫模型的比对算法[10]。然而以上算法都无法有效处理基因组规模或几百条序列以上的大数据。
随着科学技术的不断发展,生物信息数据,气象监测数据,社交网络数据呈爆炸性增长,快速有效的处理海量数据已成为急需解决的问题。Google公司提出了Map Reduce并行计算框架用于处理大规模的并行计算问题。Apache基金会基于Map Reduce框架开发出了java编程平台Hadoop[11],可以实现多机并行处理大规模数据。本文利用Map Reduce框架,结合基于k-band的多序列星比对算法[12-13],引入并行计算来处理大规模数据,实验结果表明:相对于单机处理程序,本文提出的方法能显著提高多序列比对的运算效率。
1 基于Map Reduce框架的多序列比对方法
1.1空隙罚分
由于序列比对是求解序列之间最大相似性的问题,所以需要一个判断相似性的度量方法,于是引入了空隙罚分机制。对两个或者多个字符串序列通过匹配相应的字符或者插入空格从而得到序列之间的最大相似性。虽然加入空格匹配序列的方法能够获得较好的匹配效果,但是极可能会破坏其生物学意义。空隙罚分就是用来防止无限制地引入空格,补偿插入和缺失对序列相似性的影响。对于一个空格数为p的罚分计算公式如下:

其中a表示空位罚分(gap opening),即每插入一个空隙的罚分值,b表示空位扩展罚分(gap extension),即每添加一个空格的罚分值,p为插入的空格数。
对于多序列比对假设有基因序列s={s1,s2,……sn},用score(x,y)表示两条序列x和y之间的比对得分,那么这组基因序列的比对分值spscore(s)就表示为:

1.2多序列星比对算法
动态规划算法是最常用于双序列比对的算法,即Needleman和Wunsch提出的Neeedlemana-Wunsch算法[14]。由于动态规划的计算时间复杂度为O(n^2),但是待比对的序列差异性通常不会太大,动态规划的主要回溯路径集中在二维表的主对角线上,为减小算法的时间复杂度以及空间复杂度,在计算动态规划比对算法的时候可以以主对角线为中心,取宽度为k的一条窄带上的相似性分值。对于两条长度分度分别为n,m的不等长序列,假设m>n,k的宽度初始时至少为m-n,此时最差的相似性分值为M(m-k-1)-2(k+1)(a+b),其中M为字符匹配的得分值,a为空隙罚分,b为空格罚分,如果计算出的相似性分值小于最差的相似性分值,就需要适当地扩大k的数值,从而找到更好的序列比对方案。
星比对算法就是挑选一条序列作为中心序列,用这条中心序列和其他序列依次进行比对,从而获得比对后的更新序列以及新的中心序列。对于相似性较高的基因序列,先任意选取一条序列作为中心序列,用该中心序列依次与其他序列相互比对,然后汇总更新后的中心序列,获得最终比对后的中心序列,再利用这条中心序列对第一次更新的序列进行二次修正,获得最终的更新序列组,具体计算方法如下。
星比对算法:

1.3Map Reduce的实现方法
由于Map Reduce每条记录是以(key,value)形式输入,其中key用序列名表示,value用基因序列所代表的碱基字符来表示。对于用户输入的文件可能会存在错误,如文件格式不满足要求,序列字符可能出现非法字符的情况要进行辨别与修正。首先把文件转化为适应hadoop的键值对模型,在转化文件结束后,提取序列组中的第一条序列并保存,作为中心序列。
然后进入Map Reduce框架进行序列比对计算,在第一个Map函数处理阶段,数据文件会被自动分割成一个个大小为64 M的split文件,每个Map函数在不同的Data Node节点机上执行,中心序列同时和每个Map块上的序列进行比对,每次比对会生成两条更新序列,一条是原中心序列的更新(New_center_sequence[i]),一条是序列i的更新(New_sequence[i]),然后将比对的结果保存为(sequence_name,value)的格式,其中value的值为New_center_sequence[i]与New_sequence[i],这两条序列之间用tab分隔符隔开,Map_1函数输入与输出结果,详见图1。
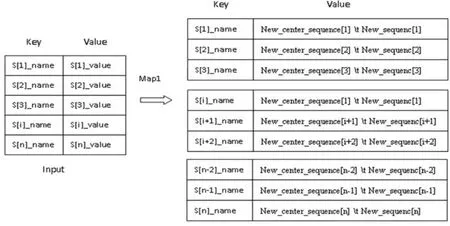
图1 第一阶段Map函数输入与输出

进入reduce阶段后,reduce函数并不对数据进行处理,直接把结果输出到HDFS文件系统下。随后从HDFS文件系统中将结果复制回本地文件系统,并从文件中分离出更新中心序列(New_center_sequence[i])。随后对更新中心序列进行汇总,汇总的方法即对于n条更新序列,统计各条序列各个空隙所具有的空格数,求出相应空隙所具有的最大空格数,即为最终中心序列(Final Center Sequence)的空隙插入方式,接着进入第二阶段Map Reduce,具体计算方法如下。
中心序列汇总:


第二阶段Map Reduce的过程基本和第一阶段相同,采用汇总后的中心序列与第一阶段输出的更新序列(New_sequence[i])再次进行比对。不同之处在于,Map函数并不保存比对后的中心序列,只保存二次比对序列i的最终更新结果(Final_new_sequence[i]),Map_2函数输入与输出,详见图2。

图2 第二阶段Map函数输入与输出
2 实验结果
实验基于Hadoop1.0版本完成,集群配置分别采用1个Name Node,2个Data Node以及1个Name Node,4个Data Node两组对比实验,用于对比硬件扩展对于运算效率的影响。同时添加了一组单机版本的实验,以观测并行计算相对于单处理在运算效率上是否有所提高。实验运行环境配置如下:单机以及集群的Name Node节点均采用64位Ubuntu操作系统,CPU为Intel(R)Core(TM)i7-3820@3.60 GHz,内存8 G*8。集群的Data Node节点采用64位Ubuntu操作系统,CPU为Intel(R)Xeon(R)E31230@3.2 GHz,内存4 G*8。
为便于比对不同数据规模对于运算性能的提升,实验采用大小分别为1 G、2 G、4 G、8 G的人类Mitochondrial DNA数据(每条序列的长度约为16 kbp)进行分组观察,统计程序5次运算时间并选取其中位数(图3),从图中可以观测得到,在相同数据规模的情况下,基于Map Reduce框架的多序列比对程序相对于单机程序所需运算时间降低,同时随着数据规模的增大,程序的运算效率进一步提高,这表明基于Map Reduce框架的多序列比对程序能很好地处理大规模数据。
由于单机处理所需运算时间的基数过大,图3所示的不同配置集群在运算效率提升上的差异性不是特别明显,由此引入加速比来衡量并行系统或程序并行化性能。加速比是同一个任务在单处理器系统和并行处理器系统中运行消耗时间的比率,计算公式如下:

图3 不同集群配置在不同数据规模上的运算时间

其中Sp为系统加速比,T1是单处理器下的运行时间,Tp是并行系统中的运行时间。统计不同配置集群的加速比结果(图4),可以直观的观测到随着硬件配置的扩展,系统运算效率的提升表现更加明显。当数据规模进一步增大时,适当的扩充现有的硬件条件,可以获取更高的运算效率。
本文提出了一种基于Map Reduce框架,结合星比对算法的多序列比对方法,通过引入并行计算来提升程序的运行效率。实验证明:随着问题规模的扩大,与单机单线程的多序列比对软件相比[1,5],基于Map Reduce的多序列比对方法能提升序列比对的运算效率,同时程序具有良好的硬件扩展能力。为了方便研究使用,作者还开发了可以在Hadoop平台上使用的多序列比对软件MSA Hadoop1.0(http://datamining.xmu.edu.cn/software/MSA Hadoop),研究者可以在上述网址中下载到本文实验中提及的软件以及操作指南。
3 多序列比对在肿瘤研究中的应用
多序列比对的结果有助于发现单核苷酸多态性(SNP),拷贝数差异(CNV)和序列缺失(indel)。而已有大量研究表明,肿瘤的产生机制,遗传效应和变异程度均与上述现象相关。2004年,日本研究者通过对人类的线粒体DNA进行测序,然后利用多序列比对的方法发现,阿兹海默症和糖尿病均与线粒体DNA上的若干敏感位点相关[15]。此外,通过多序列比对和将测序序列向基因组上回贴(mapping),也可以找出肿瘤患者与正常人在部分基因上的表达差异,相关研究发现MicroRNA的表达差异与肿瘤有密切关系[16-17]。还有研究发现,MicroRNA不但在表达量上产生差异,而且成熟体的序列上也有细微差异,这种差异被称为isomiR(miRNA的多样性表达)[18],这些isomiR的比较分析依然需要多序列比对的方法。目前在肿瘤遗传学研究中,尽管对SNP难以定义,但全基因组关联分析(GWAS)依然是主要的研究方法。从肿瘤细胞到癌旁细胞,从未转移的肿瘤细胞到转移的肿瘤细胞的对比分析依然需要依靠GWAS,而相关的测序数据不但要映射回基因组找出差异,还需要在立体的层面对所有的位点进行对齐和分析,随着GWAS研究的深入,已不仅是对SNP位点进行关联分析[19],还需要更多的情况考虑序列的插入和缺失,以及各种基因的表达量等,这对多序列比对提出了更高的挑战。因此,关于多序列比对的理论与内容还要面对更大规模的数据研究,以及融合入更多信息的复杂模型。
[1]ThomposonJD,GibsonTJ,HigginsD.CLUSTALW:improving the sensitivity of progressive multiple sequence alignment through sequence weighting position-specific gap penalties and weight matrix choice[J].Nucleic Acids Res,1994,22(22):4673-4680.
[2]Noredama C,Holm L,Higgins DG.COFFEE:an objective function for multiple sequence alignments[J].Bioinformatics,1998,14(5):407-422.
[3]Katoh K,Toh H.Parallelization of the MAFFT multiple sequence alignment program[J].Bioinformatics,2010,26(15):1899-1900.
[4]Hirosawa M,Hoshida M,Ishikawa M,et al.MASCOT: multiple alignment system for protein sequences based on three-way dynamic programming[J].Bioinformatics,1993,9(2):161-167.
[5]邹权,郭茂祖,王晓凯,等.基于关键字树的DNA多序列星比对算法[J].电子学报,2009,37(8):1746-1750.
[6]袁激光,金人超,李红涛.基于A*算法的启发式算法求解多序列比对问题[J].华中科技大学学报(自然科学版),2003,31(9):50-52.
[7]Kim J,Pramanik S,Chung MJ.Multiple sequence alignment using simulated annealing[J].Bioinformatics,1994,10(4):419-426.
[8]Notredame C,O'Brien EA,Higgins DG.RAGA:RNA sequence alignment by genetic algorithm[J].Nucleic Acids Research,1997,25(22):4570-4580.
[9]张鹏帅,霍红卫.多序列比对问题的粒子群优化算法求解[J].计算机工程与应用,2005,18:84-87.
[10]Krogh A,Brown M,Mian IS,et al.Hidden Markov models in computational biology:Applications to protein modeling[J].Journal of Molecular Biology,1994,235(5): 1501-1531.
[11]Zou Q,Li XB,Jiang WR,et al.Survey of Map Reduce frame operation in bioinformatics[J].Brief Bioinfor,2014,15(4):637-647.
[12]Zou Q,Shan X,Jiang Y.A novel center star multiple sequence alignment algorithm based on affine gap penalty and K-band[J].Physics Procedia,2012,33:322-327.
[13]Zou Q,Hu Q,Guo M,et al.HAlign:Fast multiple similar DNA/RNA sequence alignment based on the centre star strategy[J].Bioinformatics,2015,31(15):2475-2481.
[14]Needleman SB,Wunsch CD.A general method applicable to the search for similarities in the amino acid sequence of two proteins[J].J Mol Biol,1970,48(3):443-453.
[15]Tanaka M,Cabrera VM,González AM,et al.Mitochondrial genome variation in eastern Asia and the peopling of Japan[J].Genome Res,2004,14(10a):1832-1850.
[16]Zou Q,Mao Y,Hu L,et al.miRClassify:An advanced web server for miRNA family classification and annotation[J].Comput Biol Med,2014,45:157-160.
[17]Wang Q,Wei L,Guan X,et al.Briefing in family characteristics of microRNAs and their applications in cancer research[J].BBA-Proteins Proteom,2014,1844(1):191-197.
[18]Guo L,Chen F.A challenge for miRNA:multiple isomiRs in miRNAomics[J].Gene,2014,544(1):1-7.
[19]Li P,Guo M,Wang C,et al.An overview of SNP interactions in genome-wide association studies[J].Brief Funct Genomics,2015,14(2):143-155.
R730
A
10.11877/j.issn.1672-1535.2016.14.06.03
2015-10-15)
国家自然科学基金(61370010)
(corresponding author),邮箱:zouquan@nclab.net
猜你喜欢
数学物理学报(2022年5期)2022-10-09
中学生数理化(高中版.高考数学)(2022年1期)2022-04-26
小天使·三年级语数英综合(2021年3期)2021-06-15
数学小灵通(1-2年级)(2021年4期)2021-06-09
河北画报(2020年8期)2020-10-27
数学小灵通(1-2年级)(2020年6期)2020-06-24
数学小灵通(1-2年级)(2019年12期)2019-12-26
中学生数理化·八年级数学人教版(2017年2期)2017-03-25
浙江大学学报(工学版)(2016年2期)2016-06-05
俄罗斯问题研究(2013年1期)2013-03-11
