
高效液相色谱-串联质谱法快速检测海参中硝基呋喃代谢物前处理方案的探讨
2016-11-08 11:15孙稚菁张国翠任国杰卢熠川
分析测试学报 2016年9期
孙稚菁 ,王 婵,李 崇 ,张国翠 ,任国杰 ,卢熠川
(1.大连市产品质量检测研究院,辽宁 大连 116030;2.大连标准检测技术研究中心,辽宁 大连 116030)
高效液相色谱-串联质谱法快速检测海参中硝基呋喃代谢物前处理方案的探讨
孙稚菁1,王婵2*,李崇2,张国翠2,任国杰2,卢熠川2
(1.大连市产品质量检测研究院,辽宁大连116030;2.大连标准检测技术研究中心,辽宁大连116030)
利用高效液相色谱-串联质谱(HPLC-MS/MS)联合改进的QuEChERS建立了同时测定活海参中硝基呋喃类药物的代谢物氨基脲、1-氨基乙内酰脲、3-氨基-2-唑烷基酮、5-甲基吗啉-3-氨基-2-唑烷基酮的方法。样品经盐酸水解,2-硝基苯甲醛衍生,37 ℃水浴16 h,调节至pH 7.0 ~7.5,QuEChERS提取净化,氮吹至干后,用20%乙腈水定容,经C18色谱柱分离,以乙腈-0.1%甲酸溶液为流动相进行梯度洗脱,用HPLC-MS/MS以多反应监测模式进行检测。结果表明,该方法的线性范围为1.0 ~10 μg/L,4种代谢物的相关系数均不小于 0.999,检出限和定量下限分别为0.15 μg/kg 和0.5 μg/kg,在0.5,1.0,2.0,5.0 μg/kg的加标水平下,回收率为88.0%~109.6%,相对标准偏差(RSD)为3.8% ~11.0%。用此方法对大连地区200批海参进行检测,合格率为95%。该方法前处理操作简便,省时,溶剂消耗量小,可作为同时分析活海参中4种硝基呋喃类药物代谢物残留量的有效手段。
硝基呋喃代谢物;QuEChERS;海参;高效液相色谱-串联质谱
硝基呋喃类药物是一种广谱抗生素,对大多数革兰氏阳性菌和革兰氏阴性菌、真菌和原虫等病原体均有杀灭作用。近年来,为提高海参幼苗成活率,防止生病,一些养殖户在海参养殖过程中大量添加抗生素等药物[1-2]。其中,由硝基呋喃类药物导致的活海参不合格率为10%。由于硝基呋喃类药物具有潜在的“三致”作用,欧盟1995年EC/1442/95将呋喃唑酮列为不得检出药物;我国农业部在2002 年发布1号、193号、235号公告明确禁止呋喃它酮、呋喃唑酮用于所有食品动物,规定其不得检出,在2005年的560号公告中将呋喃西林、呋喃妥因列为禁用兽药[3]。
硝基呋喃类药物主要包括呋喃唑酮、呋喃西林、呋喃它酮、呋喃妥因,被摄入后在体内极不稳定,会很快代谢,其代谢产物分别为氨基脲(SEM)、1-氨基乙内酰脲(AHD)、3-氨基-2-唑烷基酮(AOZ)、5-甲基吗啉-3-氨基-2-唑烷基酮( AMOZ)[4]。硝基呋喃及其代谢产物的分子式与结构式如表1所示。Hassan等[5]建立了检测硝呋索尔等4种硝基呋喃类药物的LC-MS/MS方法,杨琳等[6]建立了水产品及其苗种中硝基呋喃代谢物的LC-MS/MS检测方法,郭无瑕等[7]利用LC-MS/MS建立了干海参中硝基呋喃代谢物残留量的检测方法,杨奕、王传现等[8-9]利用LC-MS/MS对虾中的硝基呋喃进行检测。以上文献均用盐酸水解衍生后,固相萃取小柱净化,但其操作繁琐、耗时且成本较高,所以建立一种准确快速的硝基呋喃药物的检测方法成为亟待解决的问题。本实验在对海参样品衍生后,采用改进的QuEChERS法进行净化,兼具Quick(快速)、Easy(简单)、Cheap(便宜)、Effective(高效)、Rugged(耐用)和Safe(安全)等优点,结合HPLC-MS/MS建立了硝基呋喃代谢产物的检测方法,并对200个活海参样品进行检测,为水产品中硝基呋喃代谢物快速检测新方法的建立提供了理论依据。
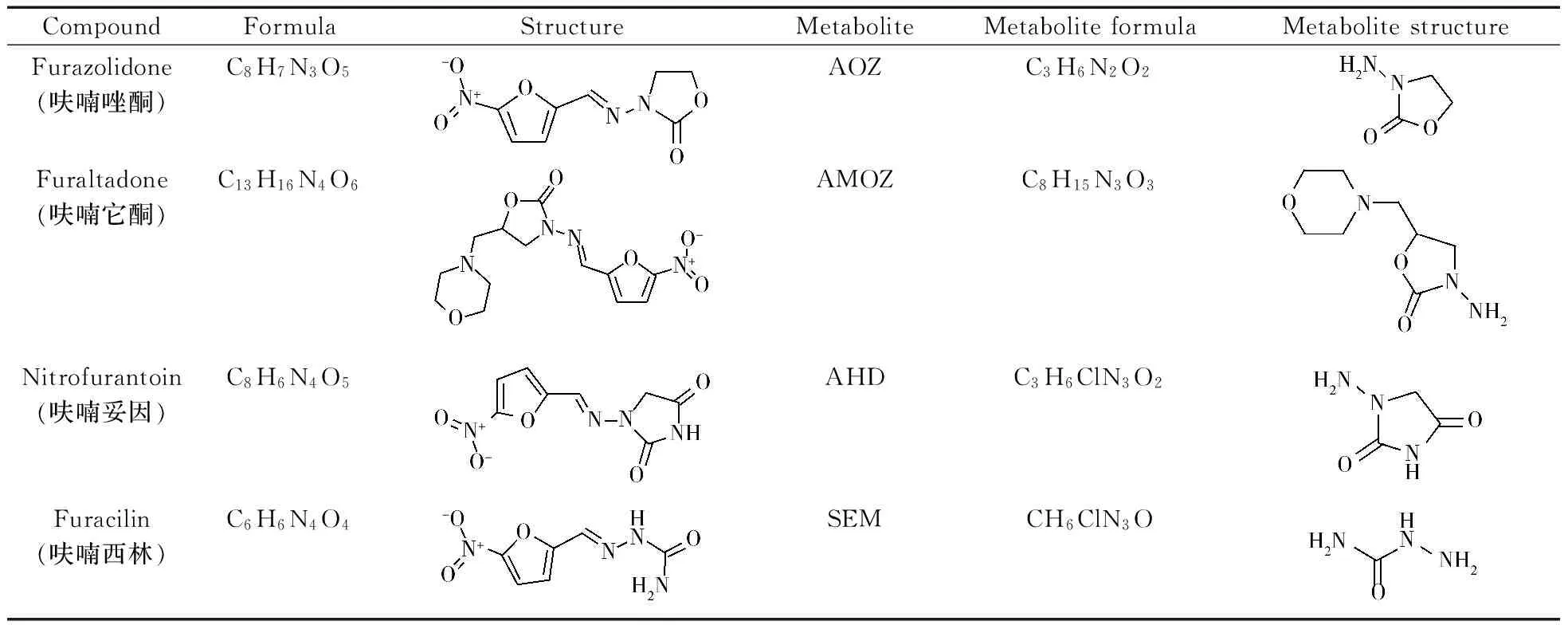
表1 硝基呋喃类药物及其代谢物的分子式、结构式Table 1 Molecular structure and structural formula of nitrofuran and its metabolites
1 实验部分
1.1仪器、材料试剂
Q-Trap 5500串联四极杆复合线性离子阱质谱仪-配电喷雾离子源(美国AB公司);旋涡混匀器(德国IKA公司);离心机(美国Sigma公司);超纯水机(美国Millipore公司)。
海参样品均为大连地区养殖海参。
乙腈(色谱纯,J&K公司);甲酸(色谱纯,迪马科技公司);Bond Elut QuEChERS萃取盐包A(内含4 g Na2SO4和1 g NaCl)、Bond Elut QuEChERS萃取盐包B(内含4 g MgSO4、1 g NaCl、1 g 柠檬酸钠、0.5 g 三水合二柠檬酸二钠) 以及Bond Elut QuEChERS净化管(1支15 mL离心管,内含50 mg PSA、150 mg C18、900 mg Na2SO4)均为美国Agilent公司产品;HCl、Na3PO4、NaOH(分析纯,科密欧试剂有限公司);2-硝基苯甲醛(纯度为98%,百灵威科技有限公司);实验用水为超纯水;硝基呋喃代谢物标准样品:AOZ、SEM(纯度≥99%,德国Dr.Ehrenstorfer GmbH),AMOZ、AHD(纯度≥99%,德国Wtega公司),内标标准样品D4-AOZ、D5-AMOZ、13C3-AHD和13C-15N2-SEM(纯度≥99%,德国Dr.Ehrenstorfer GmbH)。
准确称取 4 种硝基呋喃代谢物标准物质及其内标标准物质(精确至0.001 g),用甲醇溶解并逐级稀释定容至1.0 μg/mL,分别配制成 4 种硝基呋喃代谢物的混合标准溶液和混合内标溶液,-18 ℃避光保存,保存期6个月。
1.2实验条件
1.2.1样品处理及净化将样品搅碎捣匀,准确称取样品4.00 g(精确至0.01 g)置于50 mL具塞离心管中,向样品中加入0.1 μg/mL硝基呋喃代谢物混合内标溶液100 μL,涡旋混合1 min后加入0.2 mol/L盐酸溶液8 mL和衍生化试剂2-硝基苯甲醛0.5 mL,涡旋混合1 min后于37 ℃恒温水浴振荡16 h衍生。衍生后的样品加入1 mL 0.3 mol/L Na2SO4,用2 mol/L NaOH调至pH 7.0 ~7.5,向样品中加入10 mL乙腈,涡旋均匀后加入萃取盐包,涡旋混匀1 min后于4 ℃ 8 000 r/min离心5 min,取6 mL上清液加入净化管中净化,涡旋1 min后于4 ℃ 8 000 r/min 离心5 min,取5 mL上清液在40 ℃下氮气吹干,1 mL 20%乙腈水定容,过0.22 μm滤膜,上机测定。
1.2.2液相色谱-串联质谱条件色谱柱:Agilent Poroshell 120(3.0 mm×50 mm×2.7 μm);流动相:A为水(含0.1%甲酸),B为乙腈;柱温箱:40 ℃;流速:0.3 mL/min;进样体积:2 μL。梯度洗脱程序:0~0.5 min,18%B;0.5~4 min,18%~50%;4~4.5 min,50% B;4.5~7 min,50%~82% B。采用电喷雾离子源,喷雾电压(IS):5 500 V;雾化器压力(GS1):50 psi(344.5 kPa);气帘气压力(CUR):35 psi(241.3 kPa);辅助气压力(GS2):50 psi(344.5 kPa);离子源温度(TEM):550 ℃。定性离子对和定量离子对等参数见表2。
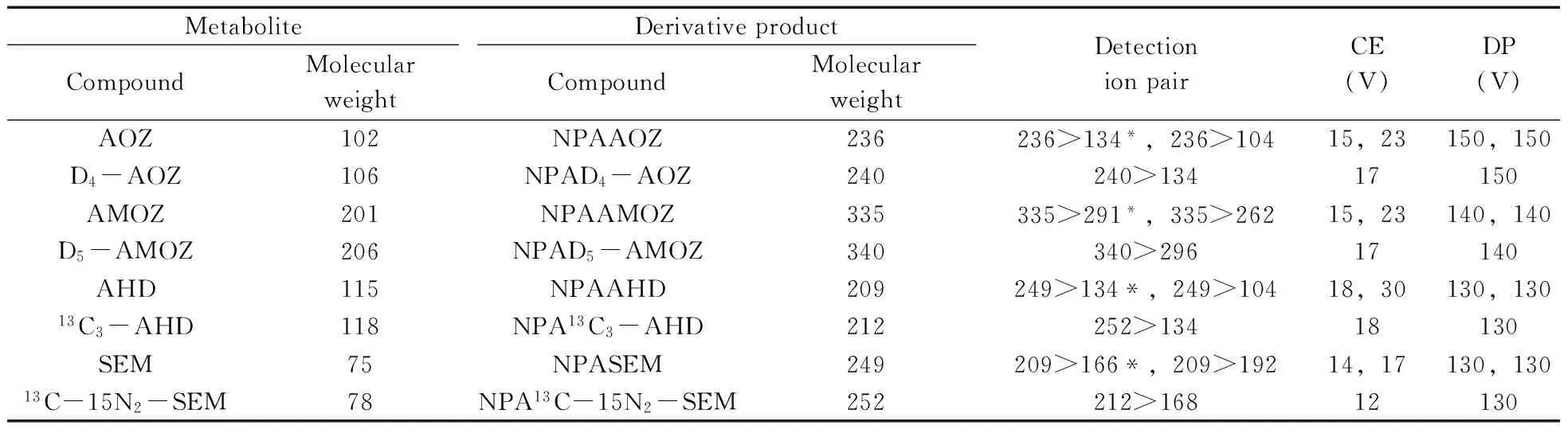
表2 代谢衍生产物的分子量及质谱参数Table 2 Molecular weight and MS parameters of nitrofurans and its metabolites
*quantitative ion
2 结果与讨论
2.1前处理条件的优化
2.1.1衍生化原理及试剂的选择硝基呋喃代谢物常用的衍生化试剂有2-硝基苯甲醛(2-NPA)[10]、2-氯苯甲醛(2-CPA)[11]、2-羟基-1-萘甲醛(2-HN)[12]等,本实验采用2-NPA作为衍生试剂,代谢物的亲核基团R-NH2在酸性催化环境很快游离出来与2-NBA碳酰基发生化学作用[13]。4种代谢物及其相应的衍生产物分别为3-[2-硝基苯(亚甲基)]-氨基-唑酮(NPAAOZ)、5-甲基吗啉-[2-硝基苯(亚甲基)]-氨基-2-唑酮(NPAAMOZ)、[2-硝基苯(亚甲基)]-氨基脲(NPASEM)和[2-硝基苯(亚甲基)]-氨基-2-内酰脲(NPAAHD)[14]。
2.1.2固相萃取与QuEChERS的比较 QuEChERS方法是由Anastassiades和Lehotay于2003年首先在EPRW会议提出,于2003年正式发表的用于农产品中多农药残留分析的前处理方法。由于动物样品中含有大量的脂肪、蛋白质等物质难以去除,可能对检测产生较大的干扰,因此QuEChERS 方法在兽药残留检测方面的应用少于农药残留。
传统的硝基呋喃代谢物检测方法一般选用固相萃取小柱净化,本实验采用改进的QuEChERS方法可降低时间和溶剂成本。将均质后的样品经乙腈提取后,采用萃取盐析分层,利用基质分散萃取机理,采用PSA 、C18、无水Na2SO4与基质中绝大部分干扰物(脂肪酸和蛋白质)结合,通过离心方式去除,从而达到净化目的。分别采用GB/T 20752-2006[15]与改进的QuEChERS法对海参样品进行加标回收实验,内标法定量。结果显示,两种前处理方法的回收率均可达到90% ~110%,且回收率差距较小,但选用固相萃取小柱操作繁琐耗时,消耗溶剂较多,因此最终选择改进的QuEChERS作为前处理方法。
2.1.3QuEChERS萃取盐包的选择 比较了“1.1”所示的两种萃取盐包的萃取效率,外标法定量。结果表明,盐包A的萃取效率比盐包B的萃取效率低5% ~17%,可能是由于MgSO4相比Na2SO4具有更大的吸水容量,能明显减少水相,从而促进待测物在有机相中的分配和两相的分层。且在萃取-分配步骤中,MgSO4水合作用是一个强放热过程,可使萃取液变热,达到40 ~45 ℃,更有利于待测物从水相到有机相的转移。在萃取液中添加柠檬酸钠和三水合二柠檬酸二钠缓冲盐,也使得提取溶液保持相对稳定的pH值。因此实验最终选择萃取盐包A。
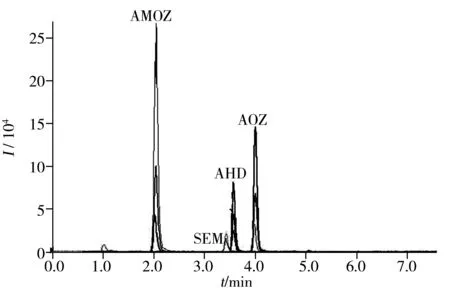
图1 4种硝基呋喃代谢产物的总离子流色谱图Fig.1 Total ion chromatogram of four kinds of nitrofuran metabolites
2.2方法验证
2.2.1标准曲线与检出限在优化实验条件下,按“1.2.1”前处理方法制备1.0,2.0,5.0,7.0,10 μg/L的系列混合标准溶液,内标法定量(内标100 μg/L) ,以分析物与内标物的峰面积比值(y) 对分析物浓度(x,μg/L) 进行线性回归,4种硝基呋喃类药物的代谢物在1.0 ~10 μg/L范围内线性良好,相关系数(r2)均不小于0.999,以3倍信噪比(S/N) 确定其检出限均为0.15 μg/kg,以10倍信噪比确定其定量下限均为0.5 μg/kg。总离子流色谱图见图1。
2.2.2回收率与精密度在海参的空白基质中分别添加4种硝基呋喃类药物的代谢物混合标准溶液及内标混合标准溶液,按“1.2.1”前处理方法进行加标回收和精密度试验,通过内标校准法计算平均回收率(n=6)和相对标准偏差(RSD,n=6),结果见表3。在0.5,1.0,2.0,5.0 μg/kg的加标水平下,4种化合物的平均回收率为88.0% ~109.6%,RSD为3.8%~11.0%,当目标化合物的检测值超出线性范围时,则稀释一定倍数使其在线性范围内后再进行定量分析。
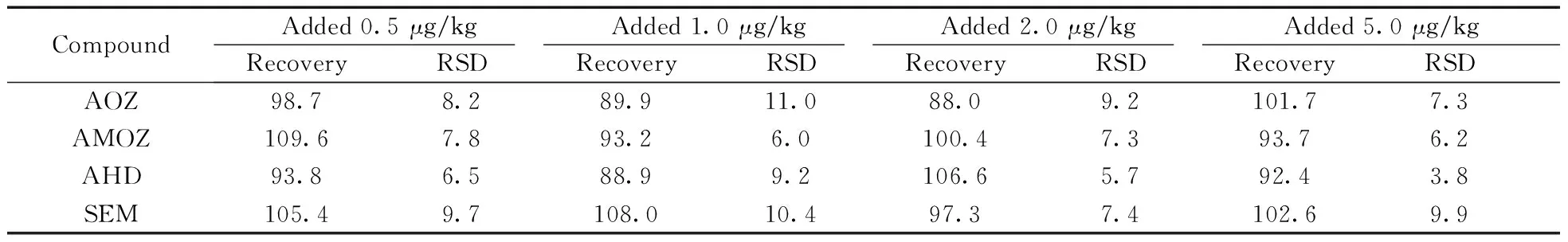
表3 4种硝基呋喃代谢产物的平均回收率及相对标准偏差(n=6,%)Table 3 Average recoveries and relative standard deviations of 4 kinds of nitrofuran metabolites(n=6,%)
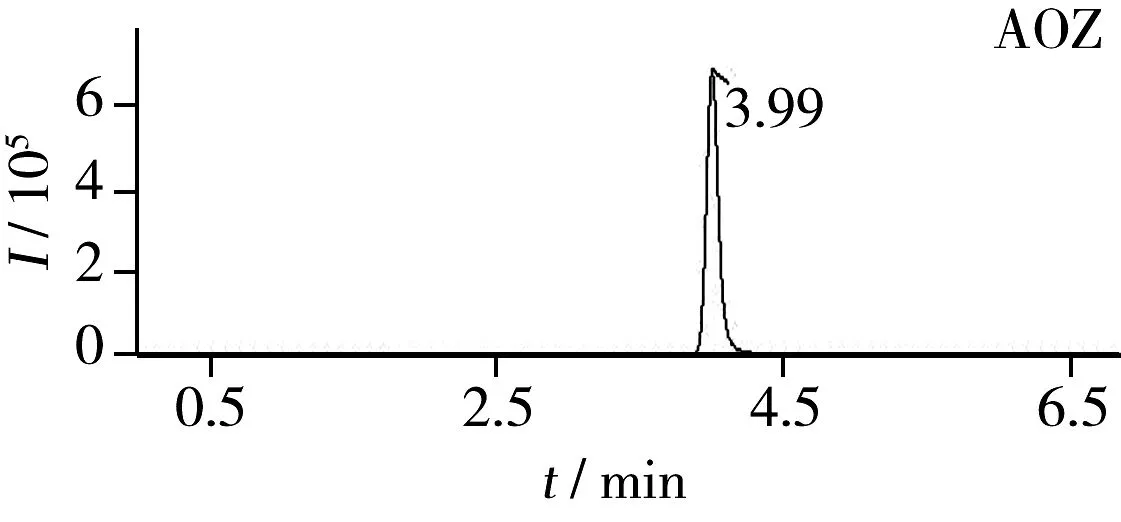
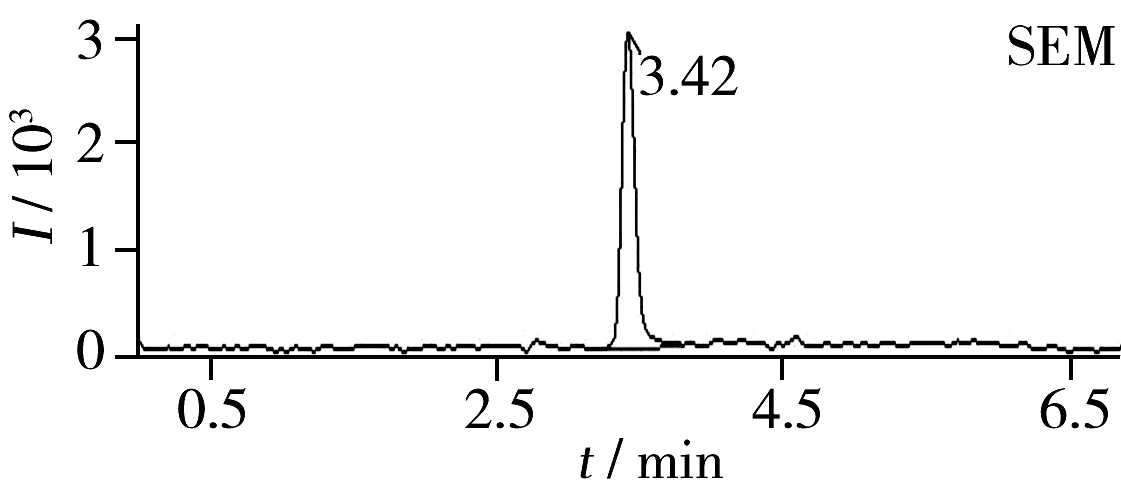
图2 海参阳性样品的TIC图Fig.2 TIC chromatograms of holothurian positive samples
2.3实际样品的测定
采用本实验建立的检测方法对200批活海参进行测定,结果显示,10批活海参检出AOZ,最大检出量为48.2 μg/kg,3批活海参检出SEM,最大检出量为6.40 μg/kg,另外两种硝基呋喃代谢物均未检出,200批活海参的合格率为95%。阳性样品的TIC图见图2。
3 结 论
本文采用改进的QuEChERS结合LC-MS/MS建立了硝基呋喃代谢物的分析方法,回收率为88.0%~109.6%,RSD为3.8%~11.0%,检出限为0.15 μg/kg。用于200批活海参的检测,合格率为95%。该方法前处理简单、重复性好、检出限低,可实现4种硝基呋喃代谢物的同时检测,完全满足国内外硝基呋喃类药物的监控和检测要求。
[1] An H,Parrales L,Wang K,Cain T,Hollins R,Forrest D,Liao B,Paek H C,Sram J.J.AOACInt.,2015,98(3):602-608.
[2] Veach B T,Baker C A,Kibbey J H,Fong A,Broadaway B J.J.AOACInt.,2015,98(3):588-594.
[3] Chen M M.StudyofNitrofuranMetabolitesDetectionMethod.Hefei:Anhui University (陈明明.一种硝基呋喃类药物代谢物检测方法的研究.合肥:安徽大学),2013:1-8.
[4] El-Demerdash A,Song F,Reel R K,Hillegas J,Smith R E.J.AOACInt.,2015,98(3):595-601.
[5] Hassan M N,Rahman M,Hossain M B,Hossaina M M,Mendesd R,Nowsada A A K M.Egypt.J.Aqua.Res.,2013,39(1):51-58.
[6] Yang L,Fu H,Liu Q.FoodSci.(杨琳,傅红.刘强.食品科学),2010,31(12):206-211.
[7] Guo W X,Li X F.J.DalianOceanUniv.(郭无瑕,李肖斐.大连海洋大学学报),2013,28(6):614-616.
[8] Yang Y,Shao B.J.FoodSaf.Qual.(杨奕,邵兵.食品安全质量检测学报),2013,4(1):135-140.
[9] Wang C X,Huang F,Wang M,Sheng Y G,Zhang J,Han L,Song Q,Li X H,Xu D M,Ding Z P.Chin.J.Chromatogr.(王传现,黄帆,王敏,盛永刚,张缙,韩丽,宋青,李晓虹,徐敦明,丁卓平.色谱),2013,31(3):206-210.
[10] Yin Y,Pan Y,Liu S G,Zheng G M,Zhu X P,Ma L S,Dai X X,Shan Q.Chin.J.Anal.Lab.(尹怡,潘宇,刘书贵,郑光明,朱新平,马丽莎,戴晓欣,单奇.分析试验室),2015,34(4):416-420.
[11] Li Y P,Lin Y H,Jia D F,Yang F,Yu K J,Liu Z C,Wang Y J,Cai C P.J.Instrum.Anal.(李耀平,林永辉,贾东芬,杨方,余孔捷,刘正才,王亚军,蔡春平.分析测试学报),2008,27(7):712-717.
[12] Du N N,Chen M M,Sheng L Q,Chen S S,Xu H J,Liu Z D,Song C F,Qiao R.J.Chromatogr.B,2014,1327:90-96.
[13] Zhu W X,Liu Y F,Liang W.Prog.Veter.Med.(祝伟霞,刘亚风,梁炜.动物医学进展),2010,31(2):99-102.
[14] Peng T,Chu X G,Yang Q,Li G,Li J Z,Li C J.Chin.J.Anal.Chem.(彭涛,储晓刚,杨强,李刚,李建中,李重九.分析化学),2005,33(8):1073-1076.
[15] GB/T 20752-2006.Method for the Determination Residues of the Metabolites of Nitrofuran in Pork,Beef,Chicken,Porcine Liver and Aquatic Products LC-MS-MS Method.National Standards of the People′s Republic of China(GB/T 20752-2006.猪肉、牛肉、鸡肉、猪肝和水产品中硝基呋喃类代谢物残留量的测定 液相色谱-串联质谱法.中华人民共和国国家标准).
Pretreatment Programs Discussion of Detecting Nitrofuran Metabolites Rapidly in Holothurians by High Performance Liquid Chromatography Tandem Mass Spectrometry
SUN Zhi-jing1,WANG Chan2*,LI Chong2,ZHANG Guo-cui2,REN Guo-jie2,LU Yi-chuan2
(1.Dalian Inspection Research Institute of Product Quality,Dalian116030,China;2.Dalian Standard Testing Technology Research Center,Dalian116030,China)
A high performance liquid chromatography-tandem mass spectrometry(HPLC-MS/MS)combined with QuEChERS method was developed for the simultaneous qualitative and quantitative analyses of four metabolites of nitrofuran antibiotics,including semicarbazide(SEM),1-amino-hydantoin(AHD),3-amino-2-oxazolidinone(AOZ),5-morpholino-3-amino-2-oxazolidone(AMOZ).The samples were hydrolyzed with HCl,and derivatized with 2-nitrobenzaldehyde at 37 ℃ for 16 h.The derivative solutions were adjusted to pH 7.0-7.5.The analytes were extracted and pured by Bond Elut QuEChERS.The separation was carried out on a C18column,using acetonitrile-0.1% formic acid as mobile phase by gradient elution.The analytes were detected by tandem mass spectrometry with electrospray ionization source under multiple reaction monitoring(MRM) mode.The method showed a good linearity between peak area ratios of the analytes to the internal standard and concentration of the analytes with correlation coefficients all above 0.999 over the dynamic range of 1.0-10 μg/L.The limits of detection(LODs) and quantitation (LOQs) of four antibiotics were 0.15 μg/kg and 0.5 μg/kg,respectively.The average recoveries of all the compounds at four spiked levels of 0.5,1.0,2.0,5.0 μg/kg ranged from 88.0% to 109.6% with RSDs (n=6) of 3.8%-11.0%.This method was used to detect 200 batches of holothurian in Dalian with the passing percentage of 95%.Compared to the traditional pretreatment method,this method is more easy to operate and less solvent comsuming,and is proved to be fast and effective for the simultaneous qualitative and quantitative analyses of the metabolites of nitrofuran antibiotics in holothurian.
nitrofuran metabolites;QuEChERS;holothurian;high performance liquid chromatography tandem mass spectrometry(HPLC-MS/MS)
2016-03-15;
2016-04-01
王婵,硕士,助理工程师,研究方向:食品安全色谱分析,Tel:0411-87923970,E-mail:wangachan37@163.com
10.3969/j.issn.1004-4957.2016.09.009
O657.63;R978.1
A
1004-4957(2016)09-1127-05
猜你喜欢
意林彩版(2022年2期)2022-05-03
化工管理(2021年7期)2021-05-13
金桥(2018年2期)2018-12-06
恋爱婚姻家庭(2018年33期)2018-07-22
中国农资(2016年1期)2016-12-01
烟草科技(2015年8期)2015-12-20
化工进展(2015年3期)2015-11-11
红蜻蜓·低年级(2014年9期)2015-03-23
无机化学学报(2014年8期)2014-02-28
无机化学学报(2014年6期)2014-02-28
