
液相色谱-质谱法测定植物源食品中氯虫苯甲酰胺残留量*
2016-11-19 07:21陈俊玉何建顺余晓薇朱惠娜
广州化工 2016年20期
陈俊玉,何建顺,余晓薇,朱惠娜
(1 东山出入境检验检疫局, 福建 漳州 363401;2 漳州出入境检验检疫局,福建 漳州 363400)
液相色谱-质谱法测定植物源食品中氯虫苯甲酰胺残留量*
陈俊玉1,何建顺2,余晓薇1,朱惠娜1
(1 东山出入境检验检疫局, 福建 漳州 363401;2 漳州出入境检验检疫局,福建 漳州 363400)
基于世界主要国家和组织对氯虫苯甲酰胺残留限量要求,选择10种代表性植物源食品作为基质,优化样品前处理方法,建立了植物源食品中氯虫苯甲酰胺残留量液相色谱串联质谱法的分析方法。方法的线性范围为2.5~50.0 μg/L,相关系数r2大于0.999;10种样品基质在0.01、0.02、 0.05 mg/kg 3个添加水平下,方法的平均回收率稳定在65.2%~107.2%之间,相对标准偏差为2.2%~9.9%,定量限为 0.01 mg/kg,本文方法灵敏、有效,适用于植物源食品中氯虫苯甲酰胺残留测定。
液相色谱-质谱法;氯虫苯甲酰胺;植物源食品
氯虫苯甲酰胺(chlorantraniliprole)是广谱杀虫剂;该类农药已在美国、加拿大、澳大利亚、我国等国家登记使用,基于安全考虑,我国在2014年8月1号实施的GB 2763-2014 食品安全国家标准-食品中农药最大残留限量中新增了氯虫苯甲酰胺的限量标准,国际食品法典委员会(CAC)、欧盟(EU)、美国和日本等各国和国际组织对氯虫苯甲酰胺最大残留量做了严格的限定[1-2]。建立高灵敏度的分析方法已十分迫切。目前已报道氯虫苯甲酰胺的检测方法主要有液相色谱法[3-5]、液相色谱-质谱联用法[6-8]、酶联免疫吸附法[9]等,植物源食品中氯虫苯甲酰胺农药残留量液相色谱-质谱法测定未见公开报道。本文探讨了干湿梅、稻米等10个不同基质中氯虫苯甲酰胺残留提取的适用性,优化了提取-净化步骤,采用乙腈为提取溶剂,以无水硫酸镁与氯化钠混合物盐析提高农药组分的萃取率,分散固相萃取,紫外检测器或二极管阵列检测器测定,建立植物源食品中氯虫苯甲酰胺残留液相色谱-质谱法分析方法。
1 实 验
1.1 仪器与试剂
液相色谱-串联质谱仪(配电喷雾离子源)或相当者;分析天平 感量0.00001 g;振荡器;旋转蒸发仪;涡旋混合器。
乙腈(色谱纯);PSA(40~63 μm);GCB(60~80目);甲醇(色谱纯);氯化钠;无水MgSO4;乙腈水溶液(准确量取10 mL乙腈和40 mL水后混合)。
氯虫苯甲酰胺标准品(Chlorantraniliprole,C18H14BrCl2N5O2,CAS编号:500008-45-7,纯度大于99%);标准储备溶液:称取10.0 mg标准品,用甲醇定容至10.0 mL,浓度为1.00 mg/mL。
1.2 样品前处理方法
1.2.1 试样制备
取有代表性样品500 g,将可食部分切碎后(不可水洗),用组织捣碎机将样品加工成浆状,混匀,装入洁净容器内,密封并标识。
1.2.2 提取及净化
称取匀质试样10.0 g,加入25 mL乙腈溶液,以15000 r/min高速匀质1 min后倒入另一50 mL离心管中。再加入约8 g氯化钠与无水硫酸镁1:1混合粉末,盖上盖子剧烈振荡2 min后,以4000 r/min的转速离心3 min。取出上清液5 mL转入装有40 mg PSA 和20 mg GCB 的50 mL 离心管中,剧烈振荡30 s 后,以4000 r/min 的转速离心3 min,移出上清液于60 ℃水浴中旋转蒸发近干,用乙腈水溶液定容至1.0 mL,供上机测定。
1.2.3 液相色谱串联质谱/质谱法测定
液相色谱条件:色谱柱:Agilent ZORBAX RRHD Eclipse Plus C18,100 mm×3.0 mm(内径),1.8 μm或相当者;流动相:A-0.1%甲酸水溶液,B-乙腈,A/B=2/8 (V/V);柱温:25 ℃;流速:0.30 mL/min;进样量:10 μL。
质谱分析条件: 质谱仪:三重四极杆质谱仪;离子源:电喷雾离子源;扫描方式:正离子;检测方式:多反应监测(MRM);干燥气温度:350 ℃;干燥气流速:9 L/min;雾化器压力:45 psi;毛细管电压:4000 V;驻留时间:200 ms;定性离子对、定量离子对、碎裂电压和碰撞能量见表1。

表1 氯虫苯甲酰胺监测定性离子对、定量离子对、碎裂电压和碰撞能量
2 结果与讨论
2.1 前处理方法的优化
2.1.1 提取和净化的选择
氯虫苯甲酰胺为中等极性化合物,水溶性较好。实验发现丙酮和乙酸乙酯提取出的提取液色素类榨汁较多,提取液颜色较深,不利于后续的净化操作;甲醇虽提取液颜色较浅但回收率低基质效应强;乙腈对于植物源食品表现除了很好的渗透性和较高的提取效率同时又对蜡类、脂肪等非极性成分的提取能力较弱。本文发现:氯化钠和无水硫酸镁均能促使水相与有机相分离。但氯虫苯甲酰胺的水溶性较大,单独加入氯化钠会损失掉其溶解在水相中的部分而使得萃取率降低。单独加入无水硫酸镁时,易板结和释放热量使农药分解。在提取液中加入氯化钠与无水硫酸镁(1:1)可以有效去除水分。
2.1.2 检测过程的净化
比较分析常用的方法液液萃取、固相萃取、分散固相萃取等几种发现:液液萃取操作较为简单、对实验条件和仪器要求不高,但是耗时长、溶剂用量大、净化效率低、易形成乳化、造成样本的损失,不便批量操作;固相萃取虽为常用的一种样品净化方法,但SPE小柱价格高、不能重复利用、耗时长;分散固相萃取可以有效达到除净化的目的。本试验中GCB对叶绿素、类胡萝卜素等杂质的净化效果非常好, PSA可以净化样品中的油脂、酯类、脂肪酸等物质。
2.2 LC-MS-MS条件的优化色谱分离条件研究
150 mm×4.6 mm,5 μm;150 mm×2.1 mm,5 μm;100 mm×2.1 mm,3.5 μm;3.0 mm×100 mm,1.8 μm的Cl8柱进行了试验比对。试验结果发现,超高压快速高分离度C18柱(3.0 mm×100 mm,1.8 μm)的峰形尖锐对称,柱子分离度显著优于其它色谱柱,能将氯虫苯甲酰胺同干扰物质完全分离,适合植物源食品基体中氯虫苯甲酰胺的残留检测。图1为氯虫苯甲酰胺的MRM色谱图。
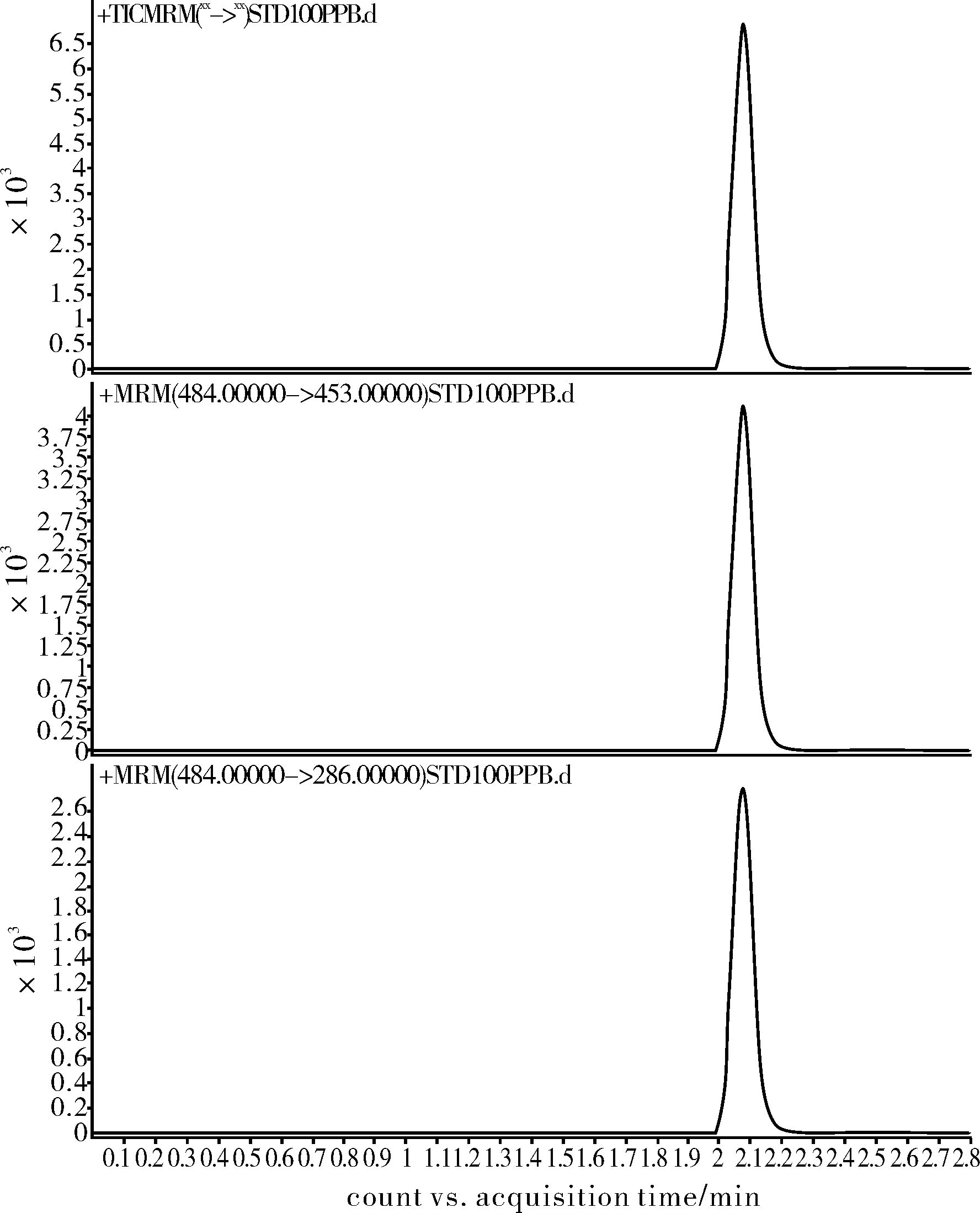
图1 氯虫苯甲酰胺液相色谱-质谱/质谱谱图
流动相的选择:实验选择甲醇-0.1%甲酸水溶液和乙腈-0.1%甲酸水溶液作为流动相体系进行色谱优化。结果表明:以乙腈做为流动相时,峰形尖锐对称,响应值较高;以甲醇做为流动相时,峰形略有拖尾,响应值较低;因此本文选用乙腈-0.1%甲酸水溶液作为流动相体系,并对流动相的比例进行优化。结果表明,当乙腈-0.1%甲酸水溶液=80:20,流速v=0.3 mL/min时,氯虫苯甲酰胺的响应值较高。

图2 氯虫苯甲酰胺MRM产物离子扫描质谱图
质谱测定条件研究 取0.2 μg/mL氯虫苯甲酰胺标准溶液进样量10 μL,进行母离子扫描,确定目标母离子。再对母离子施加一定的碰撞能量,使其产生特定的子离子。选取丰度强且干扰较少的2对子离子,以MRM模式优化各质谱参数。实验发现氯虫苯甲酰胺在正离子模式下均有响应,见图2。
2.3 方法的线性关系和测定低限
以信噪比S/N=3作为检出限,植物源性食品中氯虫苯甲酰胺的检出限为0.005 mg/kg;以信噪比S/N=10作为定量限,植物源性食品中氯虫苯甲酰胺的定量限为0.01 mg/kg。
通过对阴性样品进行加标回收实验确定的准确度和相对标准偏差。按照前面的提取净化步骤,在阴性干湿梅、黄秋葵、甘蓝、大米中添加相当于0.01,0.02, 0.05 mg/kg水平的氯虫苯甲酰胺准溶液进行添加回收试验,每个水平下测定8次。其平均回收率及相对标准偏差见表2。

表2 氯虫苯甲酰胺的平均回收率和相对标准偏差(n=8)
结果显示:在0.01,0.02, 0.05 mg/kg 3个添加水平下,方法的平均回收率稳定在66.6%~83.6%之间,相对标准偏差为2.2%~9.9%,说明方法准确度高,稳定性好。
2.4 方法的室内回收率和精密度
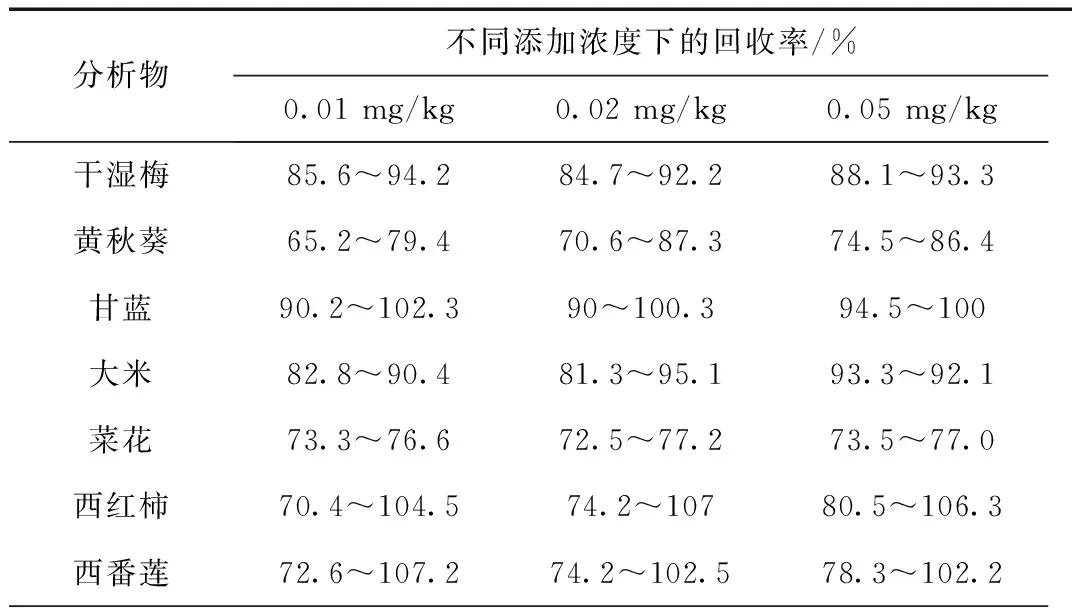
表3 氯虫苯甲酰胺的添加浓度及其回收率范围的试验数据
续表3

柑橘72.6~83.478.4~88.279.3~89.7花生71.8~97.670.2~96.378.1~102.2胡萝卜71.4~75.972.1~74.473.5~79.6
本标准方法的室内回收率和精密度实验,以不含氯虫苯甲酰胺残留的干湿梅、黄秋葵、甘蓝、大米、菜花、西红柿、西番莲、柑橘、花生、胡萝卜为空白样品基质,进行三到四个浓度水平的添加回收试验,每个浓度水平进行六次重复实验,测得各种氯虫苯甲酰胺的回收率在65.2%~107.2%范围内,其结果符合《基本规定》的要求。
3 结 论
本文选择了10种代表性的植物样品基质,选择了合适净化剂的种类及其他试剂的用量,优化了色谱分离条件,该方法适用于植物源性食品中中果蔬中甘蓝、花椰菜、叶菜类、茄果、瓜类、根茎类、薯芋类、玉米笋、柑橘类、仁果类、核果、浆果、甘蔗、大米的氯虫苯甲酰胺残留量测定。定量限为 0.01 mg/kg。方法的线性范围为2.5~50.0 μg/L,相关系数r2大于0.999;在0.01、0.02、 0.05 mg/kg 3个添加水平下,方法的平均回收率稳定在65.2%~107.2%之间,相对标准偏差为2.2%~9.9% 灵敏度满足国内外法规要求。
[1] Malhat F M. Determination of Chlorantraniliprole Residues in Grape by High-Performance Liquid Chromatography[J]. Food Analytical Methods, 2012, 5(6):1492-1496.
[2] Xu P, Ren Y, Zhou Z, et al. Determination of chlorantraniliprole in vegetables, fruits and grains by SPE clean-up and LC-UV[J]. Chromatographia, 2010, 72(7):763-766.
[3] 贺敏, 朱晓丹, 贾春虹,等. 玉米中氯虫苯甲酰胺残留的超高效液相色谱分析方法[J]. 农药学学报,2013,15(5):597-600.
[4] Zhang J M, Chai W G, Wu Y L. Residues of chlorantraniliprole in rice field ecosystem[J]. Chemosphere, 2012, 87(2):132-136.
[5] Pierluigi C, Giorgia S, Alberto A, et al. Liquid chromatography-tandem mass spectrometric ion-switching determination of chlorantraniliprole and flubendiamide in fruits and vegetables[J]. Journal of Agricultural & Food Chemistry, 2008, 56(17):7696-7699.
[6] 章虎, 钱鸣蓉, 李振,等. 高效液相色谱-串联质谱快速测定稻米中氯虫苯甲酰胺残留[J]. 农药, 2010, 49(12):906-908.
[7] 张云,郑敬峰,李耀平,等. 液相色谱-串联质谱法测定动物源性食品中氯虫苯甲酰胺和氟虫酰胺残留量[J]. 食品科学, 2013,34(10):272-275.
[8] 陈小军,王萌,范淑琴,等. QuEChERS前处理结合HPLC-MS/MS法分析氯虫苯甲酰胺在甘蓝和土壤中的残留[J]. 中国农业科学, 2012,45(13):636-264.
[9] Cui Y, Liu K, Xu C, et al. Development of a sensitive monoclonal antibody-based indirect competitive enzyme-linked immunosorbent assay for analysing chlorantraniliprole residues[J]. Food Chemistry, 2014, 143(1):293-299.
Determination of Chlorantraniliprole Pesticide Residues in Plant Source Food by Liquid Chromatogram-mass Spectrometry*
CHENJun-yu1,HEJian-shun2,YUXiao-wei1,ZHUHui-na1
(1 Dongshan Entry & Exit Inspection and Quarantine Bureau, Fujian Zhangzhou 363401;2 Zhangzhou Entry & Exit Inspection and Quarantine Bureau, Fujian Zhangzhou 363400, China)
Based on the residue limit of chlorantraniliprole by the world’s major country and organizations, 10 kinds of typical plant source food were chosen as matrix, the sample preparation methods were optimized, and the analysis method of liquid chromatogram tandem mass spectrometry to the plant source food chlorantraniliprole residue was established. The linearity range of this method was 2.5~50.0 μg/L, and correlation coefficient r2was more than 0.999. The 10 kinds of sample matrix were under three added levels of 0.01,0.02, 0.05 mg/kg. The stable average recovery rate of this method was between 65.2%~107.2%, the relative standard deviation was 2.2%~9.9%, and the limit of quantitative was 2.2%~9.9%.The method is sensitive and effective, and suitable for the determination of chlorantraniliprole residue in the plant source food.
liquid chromatogram-mass spectrometry; chlorantraniliprole; plant source food
福建检验检疫局科技项目(FK2014-DS002)。
陈俊玉(1970-),女,工程师。
O658
A
1001-9677(2016)020-0079-03
猜你喜欢
食品安全导刊(2021年21期)2021-08-30
理化检验-化学分册(2020年12期)2021-01-26
中国果菜(2020年1期)2020-02-23
农药科学与管理(2019年10期)2019-04-20
食品界(2018年8期)2018-09-03
现代农业(2016年4期)2016-02-28
中国粮油学报(2016年1期)2016-02-06
作物研究(2015年2期)2015-03-24
质谱学报(2015年5期)2015-03-01
应用化工(2014年1期)2014-08-16
