
Time representation of mitochondrial morphology and function after acute spinal cord injury
2016-12-01 12:37ZhiqiangJiaGangLiZhenyuZhangHaotianLiJiquanWangZhongkaiFanGangLv
中国神经再生研究(英文版) 2016年1期
Zhi-qiang Jia, Gang Li, Zhen-yu Zhang, Hao-tian Li, Ji-quan Wang, Zhong-kai Fan, Gang Lv
Department of Orthopedics, First Affiliated Hospital of Liaoning Medical University, Jinzhou, Liaoning Province, China
RESEARCH ARTICLE
Time representation of mitochondrial morphology and function after acute spinal cord injury
Zhi-qiang Jia, Gang Li, Zhen-yu Zhang, Hao-tian Li, Ji-quan Wang, Zhong-kai Fan*, Gang Lv*
Department of Orthopedics, First Affiliated Hospital of Liaoning Medical University, Jinzhou, Liaoning Province, China
Graphical Abstract
orcid: 0000-0001-6533-8764 (Gang Lv) 0000-0002-6927-1787 (Zhong-kai Fan)
Changes in mitochondrial morphology and function play an important role in secondary damage after acute spinal cord injury. We recorded the time representation of mitochondrial morphology and function in rats with acute spinal cord injury. Results showed that mitochondria had an irregular shape, and increased in size. Mitochondrial cristae were disordered and mitochondrial membrane rupture was visible at 2-24 hours after injury. Fusion protein mitofusin 1 expression gradually increased, peaked at 8 hours after injury, and then decreased to its lowest level at 24 hours. Expression of dynamin-related protein 1, amitochondrial fission protein, showed the opposite kinetics. At 2-24 hours after acute spinal cord injury, malondialdehyde content, cytochrome c levels and caspase-3 expression were increased, but glutathione content, adenosine triphosphate content, Na+-K+-ATPase activity and mitochondrial membrane potential were gradually reduced. Furthermore, mitochondrial morphology altered during the acute stage of spinal cord injury. Fusion was important within the first 8 hours, but fission played a key role at 24 hours. Oxidative stress was inhibited, biological productivity was diminished, and mitochondrial membrane potential and permeability were reduced in the acute stage of injury. In summary, mitochondrial apoptosis is activated when the time of spinal cord injury is prolonged.
nerve regeneration; spinal cord injury; mitochondria; fusion; fission; oxidative damage; bioenergy; mitochondrial permeability; cytochrome c; Caspase-3; apoptosis; NSFC; neural regeneration
Introduction
Spinal cord injury (SCI) has serious consequences. The main reason for the high disability rate and poor functional recovery associated with SCI is irreversible structural and functional changes in neurons induced by progressive secondary damage after the primary injury (Ambrozaitis et al., 2006; Cai et al., 2012). Complicated physiopathological mechanisms occur after SCI. Links between the various mechanisms, causes and consequences of SCI remain poorly understood. Secondary injury to subcellular organelles, such as mitochondria, has been a major focus in recent studies. Many studies have focused on the pathophysiological mechanisms in subcellular organelles after secondary injury. However, few studies have reported the effect of SCIon mitochondria. Mitochondria are “energy factories” in eukaryotic cells. Understanding the changes in mitochondrial morphology and function after secondary SCI will facilitate better understanding of the pathological mechanisms after SCI.
Mitochondria form a dynamic network and are multifunctional organelles (Bereiter-Hahn et al., 1994; Yaffe et al., 1999). The dynamic changes of mitochondria are regulated by mitochondrial dynamics proteins, including fusion protein mitofusin 1 (Mfn1), mitofusin 2 (Mfn2), optic atrophy 1, fission protein dynamin-related protein 1 (Drp1) and fission 1 (Hermann et al., 1998; Rube et al., 2004; Chan et al., 2006). The mitochondrial energy supply depends on mitochondrial fusion and fission, which contributes to information communication, including the rapid spread of membrane potential and exchange of mitochondrial contents to precisely regulate cellular activities (Rube et al., 2004; Detmer et al., 2007; Tatsuta et al., 2008). Morphological changes in mitochondria are strongly related to their function. Local ischemia, hypoxia, and changes in intracellular components after acute SCI can affect mitochondrial morphology and function. Simultaneously, mitochondria are involved in multiple mechanisms underlying the secondary injury of SCI by changing their morphology and function, including active oxygen, free radicals, calcium overload, electrolyte imbalance, and apoptosis (Abid et al., 2004; Jin et al., 2004; Giorgi et al., 2012; Solá et al., 2013).
This study investigated the changes in mitochondrial morphology and function within 24 hours after acute SCI to examine the pathological changes in secondary SCI and to lay a foundation for mitochondria-targeted therapy for SCI.
Materials and Methods
Experimental animals
This study used 108 healthy specific pathogen-free male Sprague-Dawley rats aged 10-12 years and weighing 250 ± 30 g provided by the Experimental Animal Center of Liaoning Medical University of China (license No. SCXK 2009-0004). The rats were housed in individual cages in a dry and well-ventilated room at 23-25°C, in 12-hour light/dark cycles, and allowed free access to food and water. All surgery was performed under anesthesia, and all attempts were made to minimize the pain and distress of the experimental animals. All animal experiments were carried out in accordance with the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1986). The protocols were approved by the Animal Ethics Committee of Liaoning Medical University of China.
Establishing a rat model of SCI
In this study, 108 rats were equally and randomly assigned to six groups: sham group, SCI 2-hour group, SCI 4-hour group, SCI 8-hour group, SCI 16-hour group and SCI 24-hour group. Spinal cord anterior horns of six rats per group were used for each of the following studies: observation by transmission electron microscopy, western blot assay, and biochemical tests. The rats were intraperitoneally anesthetized with 10% chloral hydrate. After disinfection, a midline dorsal incision was made to expose the T9-11spinous process and vertebral plate. The spinous process and vertebral plate were bitten with a rongeur to expose the spinal cord. In accordance with Allen’s method (Yacoub et al., 2014), a 10-g object was vertically dropped from a 6-cm height, which impacted directly on the rat spinal cord. The following responses confirmed the successful establishment of the SCI model: spinal meninges congestion, rat lower limbs affected by spasms and swinging, and rat tail swinging. In the sham group, the spinal cord at T9-11was exposed but no injury was caused.
Mitochondrial ultrastructure in rats by transmission electron microscopy
Spinal cord from T9-11segments was cut into 1-mm3blocks at corresponding time points after injury in each group. Blocks were placed in fixative at 4°C for 2 hours, washed with 0.1 M sodium dimethylarsenate, and fixed in 1% OsO4for 2 hours. The samples were dehydrated, embedded in paraffin and sliced into 50-70-nm-thick sections. These sections were placed on a copper screen, stained with uranyl acetate and lead citrate, and observed by transmission electron microscopy (Japan Electron Optics Laboratory Co., Ltd., Tokyo, Japan).
Mitochondria isolation
Spinal cord mitochondria were isolated according to mitochondria kit instructions (Applygen Technologies Inc., Beijing, China). The following method was conducted at 4°C. Spinal cord tissue (100 mg) was cut into pieces and placed in a homogenizer. Then, 1.5 mL of precooling Mito Solution was added. After triturating 20 times, the homogenate was centrifuged in a centrifuge tube at 800 × g for 5 minutes at 4°C. The supernatant was removed into an additional centrifuge tube, and centrifuged at 800 × g for 5 minutes at 4°C. The supernatant was placed into another centrifuge tube, and centrifuged at 10,000 × g for 10 minutes at 4°C. Mitochondrial deposits were present at the bottom of the tube. The supernatant consisted of cytoplasm and was used for control experiments. Then, 0.2 mL of Mito Solution was added to the tube, mitochondrial deposits were resuspended, and it was centrifuged at 12,000 × g for 10 minutes at 4°C. Mitochondria deposits were present at the bottom of the tube.
Detection of mitochondrial membrane potential

Figure 1 Mitochondrial morphology in the acute stages of SCI (transmission electron microscopy; × 10,000).
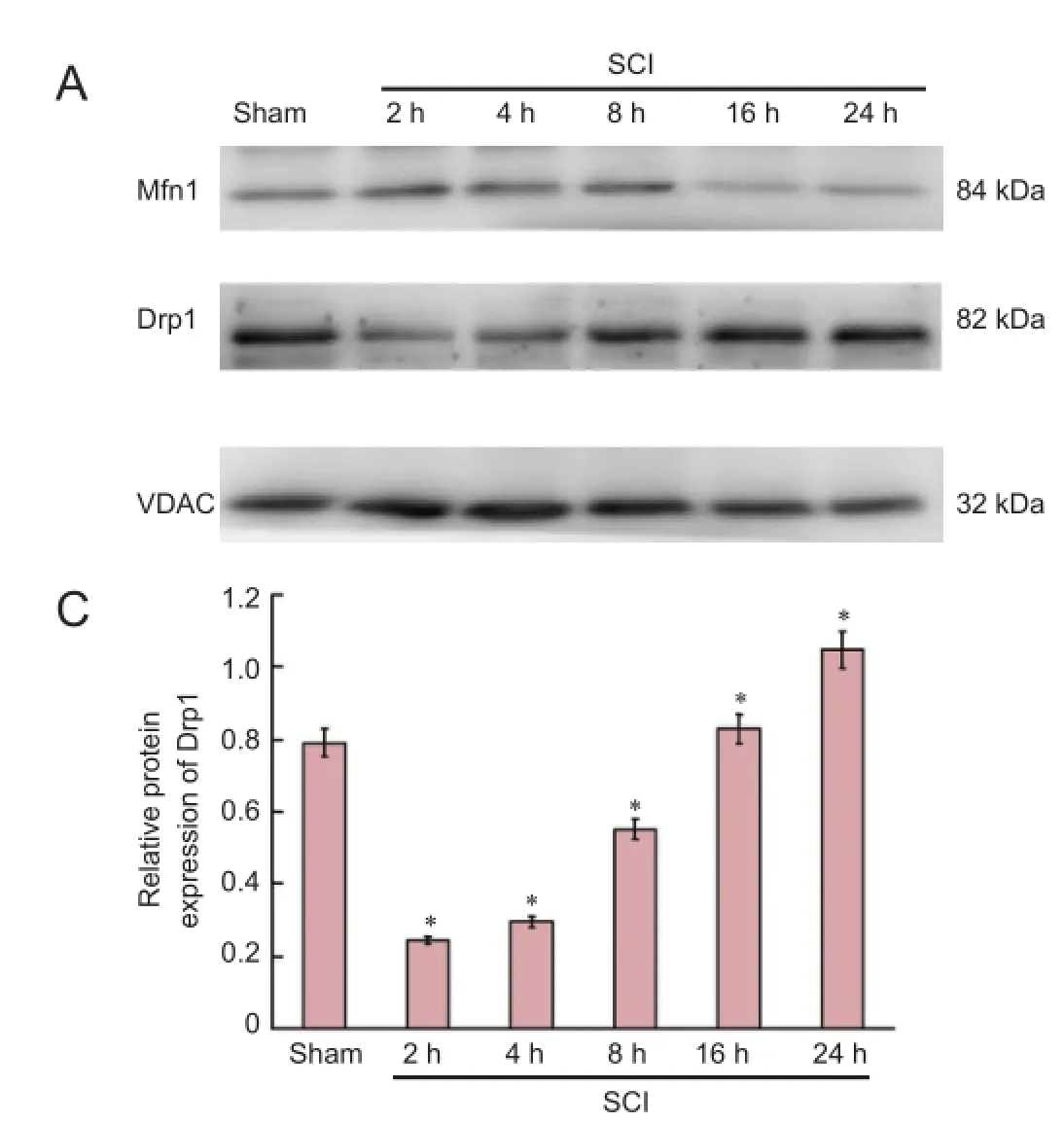
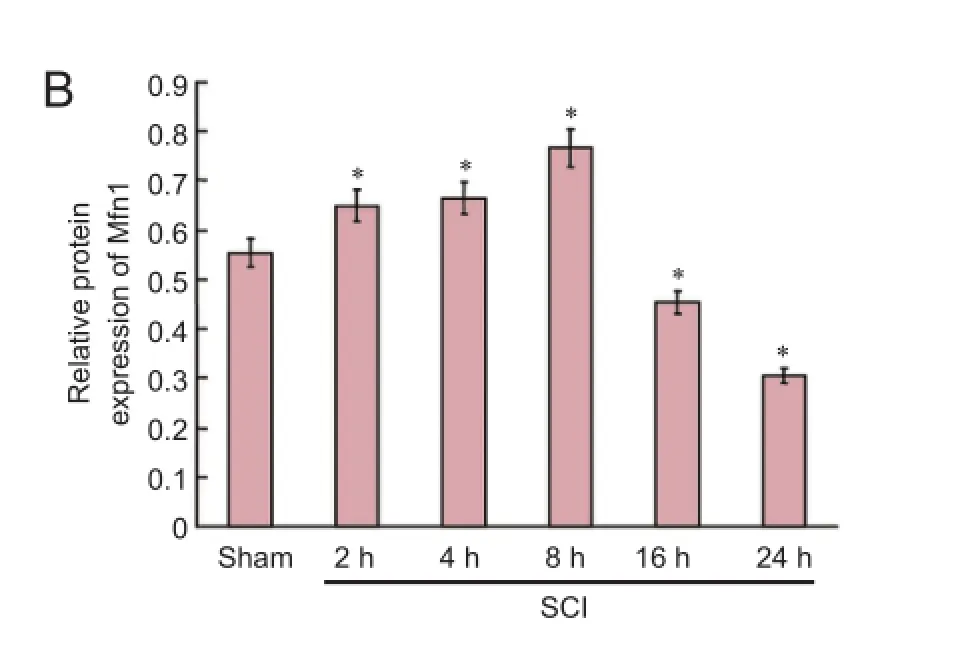
Figure 2 Expression of mitochondrial dynamics protein Mfn1 and Drp1 in the acute stages of SCI (western blot assay).
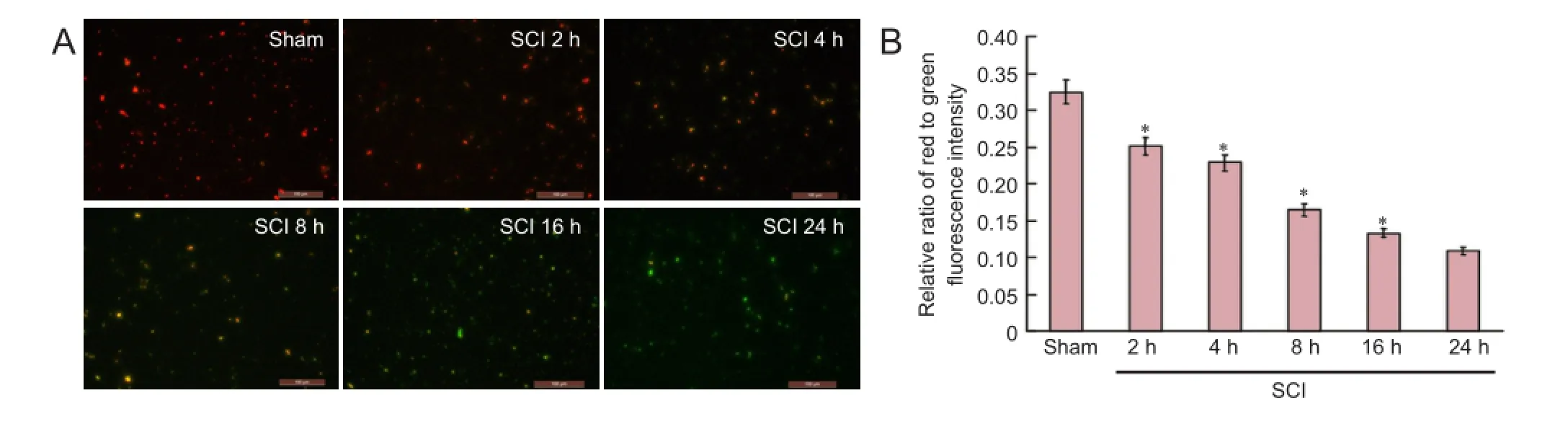
Figure 3 Changes in mitochondrial membrane potential in the spinal cord during the acute stages of SCI (immunofluorescence staining; × 200).
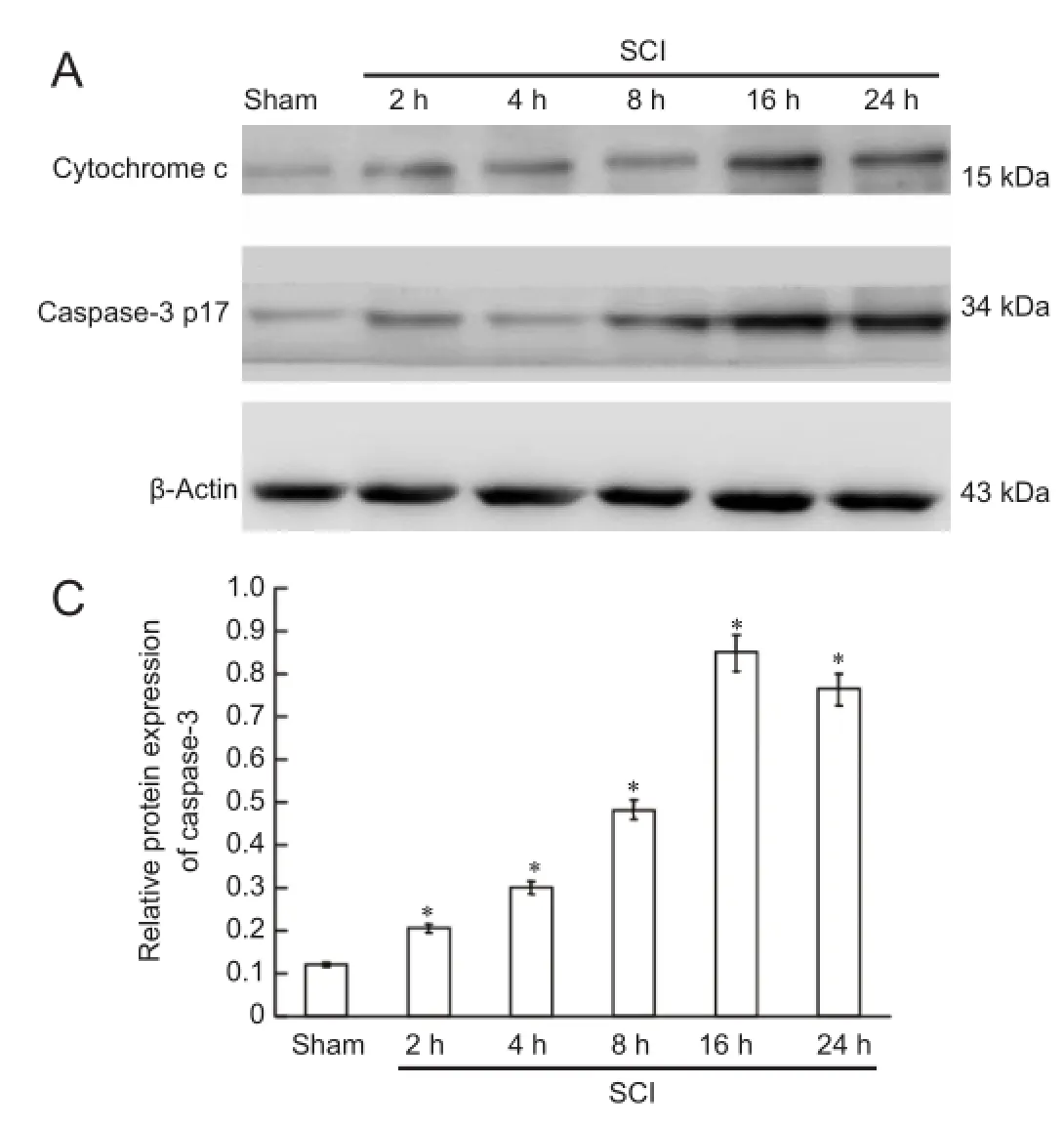

Table 1 Na+-K+-ATPase activity, malondialdehyde, glutathione and ATP contents in mitochondria during the acute stages of SCI
Mitochondria deposits were resuspended with Mito Solution at corresponding time points after injury in each group in accordance with the instructions of mitochondrial membrane potential kit (JC-1) (Beyotime Biotechnology, Haimen, China). The samples were divided into two portions for observation by fluorescence microscopy and measurement by fluorescence spectrophotometry. When the mitochondrial membrane potential was high, JC-1 in the mitochondrial matrix formed J-aggregates, which showed as red fluorescence. When the mitochondrial membrane potential was low, JC-1 did not aggregate in the mitochondrial matrix and existed as a monomer producing green fluorescence. Therefore, fluorescence microscopy (Olympus, Tokyo, Japan) was used to observe red and green fluorescence. In addition, fluorescence spectrophotometry (Shimadzu, Japan) was used to record the intensity of red and green fluorescence. To determine the fluorescence intensity, JC-1 monomers were detected under excitation wavelength of 490 nm and emitted wavelength of 530 nm and J-aggregates of JC-1 were detected under an excitation wavelength of 525 nm and emitted wavelength of 590 nm. The relative proportion of red and green fluorescence was used to determine the proportion of mitochondrial depolarization.
Determination of biological oxidation of mitochondria
Biological oxidation of mitochondria was determined using a glutathione kit, a malondialdehyde (MDA) kit, an adenosine triphosphate (ATP) kit, and a Na+-K+-ATPase kit according to the manufacturer’s instructions. All kits were purchased from the Nanjing Jiancheng Bioengineering Institute (Nanjing, China).
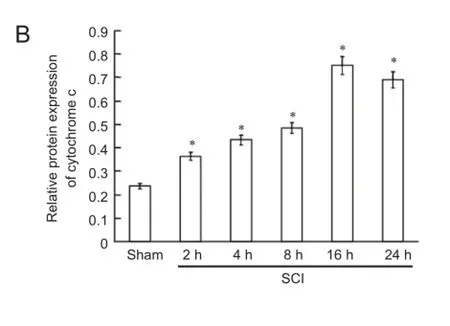
Figure 4 Cytochrome c release and caspase-3 expression in the spinal cord during the acute stages of SCI (western blot assay).
Measurement of Mfn1, Drp1, cytochrome c and caspase-3 expression in the spinal cord by western blot assay
Mitochondrial suspensions were lysed with lysate to prepare mitochondrial proteins. The samples were treated with protease inhibitors and maintained at −20°C. Cytosolic proteins were prepared from mitochondria isolated with a mitochondria isolation kit. Protein contents were determined using the bicinchoninate acid assay. Proteins were electrophoresed by 10%, 12% and 15% sodium dodecyl sulfate-polyacrylamide gels, then transferred to polyvinylidene difluoride membranes by the wet transfer method. Membranes were washed with Tris-buffered saline containing Tween 20 (TBST), blocked with bovine serum albumin at room temperature for 1 hour, and incubated with primary antibodies at 4°C overnight (mouse anti-Mfn1 monoclonal antibody, 1:1,000, Abcam, Cambridge, UK; rabbit anti-Drp1 monoclonal antibody, 1:1,000, Cell Signaling Technology, USA; rabbit anti-Vdac monoclonal antibody, 1:1,000, Cell Signaling Technology, USA; mouse anti-cytochrome c monoclonal antibody, 1:1,000, Santa Cruz Biotechnology, Santa Cruz, CA, USA; rabbit anti-caspase-3 polyclonal antibody, 1:1,000, Santa Cruz Biotechnology; and mouse anti-β-actin monoclonal antibody, 1:1,000, Santa Cruz Biotechnology). After washing with TBST, the membranes were illuminated with enhanced chemiluminescence substrate (Millipore, Billerica, MA, USA), and photographed with a gel imaging system (UVP LLC, Upland, CA, USA). Semiquantitative analysis was performed using Image J software (National Institutes of Health, Bethesda, MD, USA). To determine the expression level of the protein, the grayscalevalue of Mfn1 and Drp1 was normalized to the values of the corresponding VDAC band, and the grayscale value of cytochrome c and caspase-3 was normalized to the values of the corresponding β-actin band. The experiments were performed six times.
Statistical analysis
Data were analyzed using SPSS 13.0 software (SPSS, Chicago, IL, USA). Measurement data were expressed as the mean ± SD. Comparison among multiple groups was tested by oneway analysis of variance and the least significant difference test. The α level was set at 0.05.
Results
Mitochondrial morphology in the spinal cord after acute SCI
Transmission electron microscopy demonstrated that mitochondria were regular with distinct cristae in the sham group. Mitochondria had a regular shape but were larger in the SCI 2-hour group compared with the sham group. Mitochondria were large and with disordered cristae in the SCI 4-hour group. Mitochondrial volume was increased, and mitochondrial cristae were swollen in the SCI 8-hour group. In the SCI 16-hour group, the mitochondrial structures were not distinct, mitochondrial cristae were disrupted and disordered and partial mitochondrial membrane rupture and vacuolization were visible. Different mitochondrial size, irregular mitochondria, partial mitochondrial membrane rupture and vacuolization were detected in the SCI 24-hour group (Figure 1).
Expression of mitochondrial dynamic protein Mfn1 and Drp1 in the spinal cord after acute SCI
Western blot assay revealed that mitochondrial fusion protein expression was increased, peaked at 8 hours after injury, and then decreased. Mitochondrial fission protein expression diminished, then increased, and peaked at 24 hours (Figure 2).
Biological oxidation, bioenergy and enzyme activity on the mitochondrial membrane in the acute stage of SCI
Compared with the sham group, MDA content, glutathione content, ATP content, and Na+-K+-ATPase activity were reduced in the SCI groups compared with the sham group (P< 0.05). With prolonged time of injury, antioxidant levels decreased, oxidative damage gradually occurred, mitochondrial capacity was blocked, and ATP production gradually diminished, resulting in a gradual reduction of enzyme activity (Table 1).
Changes in mitochondrial membrane potential in the spinal cord in the acute stage of SCI
Fluorescence microscopy and fluorescence spectrophotometry demonstrated that the intensity of red/green fluorescence in the SCI groups was significantly lower than that in the sham group (P < 0.01). With time prolonged, the intensity of red/green fluorescence was reduced, indicating a downward trend of the mitochondrial membrane potential (Figure 3).
Cytochrome c release and caspase-3 expression in the spinal cord in the acute stage of SCI
Western blot assay demonstrated that cytochrome c was released, and that active caspase-3 (caspase-3 p17) was expressed at 2 hours after SCI. Cytochrome c release gradually increased and caspase-3 p17 expression increased with time, peaking at 16 hours after SCI (Figure 4).
Discussion
The dynamic equilibrium between mitochondrial fusion and fission maintains the mitochondrial morphology in cells. These dynamic changes respond differently to the physiological needs of cells, such as apoptosis and cell repair (Rube et al., 2004; Tanaka et al., 2008; Shields et al., 2015; Song et al., 2015). Mfn1/2 plays a key role in mitochondrial fusion (Rojo et al., 2002). Mitochondrial fusion can be impacted in cells lacking Mfn1 or Mfn2 and mitochondrial fragmentation occurs affecting the tubular network structure, which can impact mitochondrial functions (Wong et al., 2000; Chen et al., 2003; Pyakurel et al., 2015). Drp1 is a fundamental component of mitochondrial fission, mainly localized in the cytoplasm (Smirnova et al., 2001). Drp1 assembles around the mitochondrial tubule to form a ring-like structure, altering the inter molecular distance or angle through GTP hydrolysis, gradually compressing until mitochondrial rupture occurs, producing two independent mitochondria. After mitochondrial rupture, Drp1 returns to the cytoplasm to regulate mitochondrial fission (Labrousse et al., 1999). Mitochondrial fusion and fission are not only closely related to mitochondrial morphology, but also to cell function and apoptosis (Tanaka et al., 2008). Overexpressed molecules that mediate mitochondrial fusion can promote mitochondrial fusion and inhibit apoptosis. Furthermore, the mitochondrial-network structure can disappear after fusion protein expression is down-regulated, enhancing cell sensitivity to apoptosis (Olichon et al., 2003; Lee et al., 2004; Wang et al., 2009). Overexpressed Drp1 promotes mitochondrial fission, tubular network structure rupture, and increased apoptosis (Figueroa-Romero et al., 2009). Inhibiting fission proteins suppresses apoptosis (James et al., 2003). The above studies suggest that mitochondrial fusion/fission is strongly associated with defense mechanisms or apoptotic mechanisms in cells. The precise molecular mechanisms of how mitochondrial fusion/fission contributes to cell death remain poorly understood, although mitochondrial dynamics have an important role in cell repair and apoptosis. Our results demonstrated that in the acute stage of SCI, mitochondrial fusion was increased, and then decreased; whereas mitochondrial fission was decreased, and then increased. In SCI, fusion plays a key role in the early stages and fission plays a key role in the late stages. These changes in mitochondrial dynamics cause alterations in mitochondrial morphology in the acute stage of SCI. Electron microscopy demonstrated that mitochondrial volume and cristae had changed at 4-8 hours after SCI. With prolonged injury time, mitochondrial membrane rupture and vacuolization occurred.
Oxygen free radicals, mainly produced from mitochondria, are generated during biological oxidation. The peroxidation of mitochondrial membranes affects the integrity of the structure and function of mitochondrial membranes, physiological activity of cells, and causes cellular apoptosis (Azbill et al., 1997; Kowaltowski et al., 1999; Diao et al., 2012). MDA is an endogenous genotoxic product of oxygen radical-induced lipid peroxidation. Glutathione content can indirectly reflect the ability of a body to scavenge free radicals. Therefore, determining MDA and glutathione content can indirectly indicate the generation of oxygen free radicals and the degree of tissue damage in cells (Lakroun et al., 2015). Under normal physiological conditions, the small amount of oxygen free radicals produced by mitochondria are scavenged by the scavenging system involving superoxide dismutase and glutathione to maintain the dynamic equilibrium between free radical production and scavenging. However, during secondary ischemia and hypoxia after SCI, this dynamic equilibrium is altered. The production of oxygen free radical exceeds the scavenging ability, resulting in oxygen free radical accumulation. This oxidative stress can cause cell apoptosis (Das et al., 2012). Normal mitochondrial membrane potential is required for biological oxidation and ATP production, and is necessary to maintain the physiological functions of mitochondria (Kroemer et al., 1997). Mitochondrial membrane potential is also strongly associated with apoptosis (Jiang et al., 2013; Suzuki et al., 2013). A decrease in mitochondrial membrane potential can lead to a series of changes the in inner and outer mitochondrial membrane, such as altering mitochondrial membrane permeability, releasing cytochrome C, activating Bcl-2 and caspase family members leading to apoptosis (Bernardi et al., 1999; Choi et al., 2007; Ravindran et al., 2011). In this study, MDA content was slightly increased, glutathione content was slightly decreased, and ATP content slightly reduced in the early stages after SCI. With prolonged injury time, these findings rapidly worsened. The ATP content was reduced and the Na+-K+-ATPase activity was diminished. This caused the mitochondrial ion exchange to be disordered, a decrease in mitochondrial membrane potential and mitochondrial membrane permeability, release of cytochrome C and activation of caspase-3, finally resulting in apoptosis.
Our results confirmed that mitochondrial fusion played a key role in the early stage of SCI, when changes in mitochondrial function are small; however, when mitochondrial fission played a key role, changes in mitochondrial functions were more significant. We presumed that mitochondria initiated defense mechanisms in the early stages of SCI to reduce potential pathological injury. With prolonged injury time, the ratio of mitochondrial fusion and fission changed, and the above defense mechanisms were reduced. Moreover, the activation of the mitochondrial apoptotic pathway through pathological mechanisms finally induced apoptosis. Within 4-8 hours after injury, various factors were changed, consistent with previous results (Azbill et al., 1997; Cai et al., 2006; Soane et al., 2007), suggesting the necessity of effective therapy before the cascade reaction. These findings verified that the optimal therapeutic window was less than 6-8 hours after SCI.
This study reported the pathophysiological changes caused by secondary injury with regards to subcellular organelles, recorded the time-related changes in mitochondrial morphology and function after acute SCI, laid the foundation for further study of the effects of mitochondria on SCI, provided a theoretical basis for mitochondria-targeted therapy for SCI, and provided a new target for the prevention and treatment of SCI.
Acknowledgments: We are very grateful to Jing Bi from the Department of Neurobiology, School of Basic Medical Sciences, Liaoning Medical Universty, China for technical guidance.
Author contributions: ZQJ participated in study concept and design, wrote the paper, provided and ensured the integrity of the data. GL and ZKF designed, conceived and oversaw the study, obtained the funding, and provided technical support. GL and ZYZ ensured the integrity of the data. HTL and JQW analyzed the data. All authors approved the final version of the paper.
Conflicts of interest: None declared.
Plagiarism check: This paper was screened twice using Cross-Check to verify originality before publication.
Peer review: This paper was double-blinded and stringently reviewed by international expert reviewers.
Abid MR, Schoots IG, Spokes KC, Wu SQ, Mawhinney C, Aird WC (2004) Vascular endothelial growth factor-mediated induction of manganese superoxide dismutase occurs through redox-dependent regulation of forkhead and IkappaB/NF-kappaB. J Biol Chem 279:44030-44038.
Ambrozaitis KV, Kontautas E, Spakauskas B, Vaitkaitis D (2006) Pathophysiology of acute spinal cord injury. Medicina 42:255-261.
Azbill RD, Mu X, Bruce-Keller AJ, Mattson MP, Springer JE (1997) Impaired mitochondrial function, oxidative stress and altered antioxidant enzyme activities following traumatic spinal cord injury. Brain Res 765:283-290.
Bereiter-Hahn J, Voth M (1994) Dynamics of mitochondria in living cells: shape changes, dislocations, fusion, and fission of mitochondria. Microsci Res Tech 27:198-219.
Bernardi P, Scorrano L, Colonna R, Petronilli V, Di Lisa F (1999) Mitochondria and cell death. Mechanistic aspects and methodological issues. Eur J Biochem 264:687-701.
Cai WH, Zhang N, Fang WM, Hu ZY, Jin ZS, Jia LS, Ye XJ (2006) Changes of spinal cord mitochondrial respiratory function and the contents of intramitochondria free calcium in experimental spinal cord injury model. Nanjing Yike Daxue Xuebao 26:407-409.
Cai Y, Li J, Yang S, Li P, Zhang X, Liu H (2012) CIBZ, a novel BTB domain-containing protein, is involved in mouse spinal cord injury via mitochondrial pathway independent of p53 gene. PLoS One 7:e33156.
Chan DC (2006) Mitochondrial fusion and fission in mammals. Annu Rev Cell Dev Biol 22:79-99.
Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC (2003) Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol 160:189-200.
Choi JS, Shin S, Jin YH, Yim H, Koo KT, Chun KH, Oh YT, Lee WH, Lee SK (2007) Cyclin-dependent protein kinase 2 activity is required for mitochondrial translocation of Bax and disruption of mitochondrial transmembrane potential during etoposide-induced apoptosis. Apoptosis 12:1229-1241.
Das J, Ghosh J, Manna P, Sil PC (2012) Taurine protects rat testes against doxorubicin- induced oxidative stress as well as p53, Fas and caspase 12-mediated apoptosis. Amino Acids 42:1839-1855.
Detmer SA, Chan DC (2007) Functions and dysfunctions of mitochondrial dynamics. Nat Rev Mol Cell Biol 8:870-879.
Diao L, Mei Q, Xu JM, Liu XC, Hu J, Jin J, Yao Q, Chen ML (2012) Rebamipide suppresses diclofenac-induced intestinal permeability via mitochondrial protection in mice. World J Gastroenterol 18:1059-1066.
Figueroa-Romero C, Iñiguez-Lluhí JA, Stadler J, Chang CR, Arnoult D, Keller PJ, Hong Y, Blackstone C, Feldman EL (2009) SUMOylation of the mitochondrial fission protein Drp1 occurs at multiple nonconsensus sites within the B domain and is linked to its activity cycle. FASEB J 23:3917-3927.
Giorgi C, Baldassari F, Bononi A, Bonora M, De Marchi E, Marchi S, Missiroli S, Patergnani S, Rimessi A, Suski JM, Wieckowski MR, Pinton P (2012) Mitochondrial Ca2+and apoptosis. Cell Calcium 52:36-43.
Hermann GJ, Shaw JM (1998) Mitochondrial dynamics in yeast. Annu Rev Cell Dev Biol 14:265-303.
James DI, Parone PA, Mattenberger Y, Martinou JC (2003) hFis1, a novel component of the mammalian mitochondrial fission machinery. J Biol Chem 278:36373-36379.
Jiang L, Liu Y, Ma MM, Tang YB, Zhou JG, Guan YY (2013) Mitochondria dependent pathway is involved in the protective effect of bestrophin-3 on hydrogen peroxide-induced apoptosis in basilar artery smooth muscle cells. Apoptosis 18:556-565.
Jin Y, McEwen ML, Nottingham SA, Maragos WF, Dragicevic NB, Sullivan PG, Springer JE (2004) The mitochondrial uncoupling agent 2,4-dinitrophenol improves mitochondrial function, attenuates oxidative damage, and increases white matter sparing in the contused spinal cord. J Neurotrauma 21:1396-1404.
Kowaltowski AJ, Vercesi AE (1999) Mitochondrial damage induced by conditions of oxidative stress. Free Radic Biol Med 26:463-471.
Kroemer G, Zamzami N, Susin SA (1997) Mitochondrial control of apoptosis. Immunol Today 18:44-51.
Labrousse AM, Zappaterra MD, Rube DA, van der Bliek AM (1999) C. elegans dynamin-related protein DRP-1 controls severing of the mitochondrial outer membrane. Mol Cell 4:815-826.
Lakroun Z, Kebieche M, Lahouel A, Zama D, Desor F, Soulimani R (2015) Oxidative stress and brain mitochondria swelling induced by endosulfan and protective role of quercetin in rat. Environ Sci Pollut Res Int 22:7776-7781.
Lee YJ, Jeong SY, Karbowski M, Smith CL, Youle RJ (2004) Roles of the mammalian mitochondrial fission and fusion mediators Fis1, Drp1, and Opa1 in apoptosis. Mol Biol Cell 15:5001-5011.
Olichon A, Baricault L, Gas N, Guillou E, Valette A, Belenguer P, Lenaers G (2003) Loss of OPA1 perturbates the mitochondrial inner membrane structure and integrity, leading to cytochrome c release and apoptosis. J Biol Chem 278:7743-7746.
Pyakurel A, Savoia C, Hess D, Scorrano L (2015) Extracellular regulated kinase phosphorylates mitofusin 1 to control mitochondrial morphology and apoptosis. Mol Cell 58:244-254.
Ravindran J, Gupta N, Agrawal M, Bala Bhaskar AS, Lakshmana Rao PV (2011) Modulation of ROS/MAPK signaling pathways by okadaic acid leads to cell death via, mitochondrial mediated caspase-dependent mechanism. Apoptosis 16:145-161.
Rojo M, Legros F, Chateau D, Lombès A (2002) Membrane topology and mitochondrial targeting of mitofusins, ubiquitous mammalian homologs of the transmembrane GTPase Fzo. J Cell Sci 115:1663-1674.
Rube DA, van der Bliek AM (2004) Mitochondrial morphology is dynamic and varied. Mol Cell Biochem 256-257:331-339.
Shields LY, Kim H, Zhu L, Haddad D, Berthet A, Pathak D, Lam M, Ponnusamy R, Diaz-Ramirez LG, Gill TM, Sesaki H, Mucke L, Nakamura K (2015) Dynamin-related protein 1 is required for normal mitochondrial bioenergetic and synaptic function in CA1 hippocampal neurons. Cell Death Dis 6:e1725.
Smirnova E, Griparic L, Shurland DL, van der Bliek AM (2001) Dynamin-related protein Drp1 is required for mitochondrial division in mammialan cells. Mol Biol Cell 12:2245-2256.
Soane L, Kahraman S, Kristian T, Fiskum G (2007) Mechanisms of impaired mitochondrial energy metabolism in acute and chronic neurodegenerative disorders. J Neurosci Res 85:3407-3415.
Solá S, Morgado AL, Rodrigues CM (2013) Death receptors and mitochondria: two prime triggers of neural apoptosis and differentiation. Biochim Biophys Acta 1830:2160-2166.
Song M, Dorn GW 2nd (2015) Mitoconfusion: noncanonical functioning of dynamism factors in static mitochondria of the heart. Cell Metab 21:195-205.
Suzuki Y, Hasegawa H, Tsuji T, Tsuruda K, Sasaki D, Ishihara K, Nagai K, Yanagihara K, Yamada Y, Kamihira S (2013) Relationships of diverse apoptotic death process patterns to mitochondrial membrane potential (Δψ(m)) evaluated by three-parameter flow cytometric analysis. Cytotechnology 65:59-70.
Tanaka A. Youle RJ (2008) A chemical inhibitor of DRP1 uncouples mitochondrial fission and apoptosis. Mol Cell 29:409-410.
Tatsuta T, Langer T (2008) Quality control of mitochondria: protection against neurodegeneration and ageing. EMBO J 27:306-314.
Wang X, Su B, Lee HG, Li X, Perry G, Smith MA, Zhu X (2009) Impaired balance of mitochondrial fission and fusion in Alzheimer’s disease. J Neurosci 29:9090-9103.
Wong ED, Wagner JA, Gorsich SW, McCaffery JM, Shaw JM, Nunnari J (2000) The dynamin-related GTPase, Mgm1p, is an intermembrane space protein required for maintenance of fusion competent mitochondria. J Cell Biol 151:341-352.
Yacoub A, Hajec MC, Stanger R, Wan W, Young H, Mathern BE (2014) Neuroprotective effects of perflurocarbon (Oxycyte) after contusive spinal cord injury. J Neurotrauma 31:256-267.
Yaffe MP (1999) The machinery of mitochondrial inheritance and behavior. Science 283:1493-1497.
Copyedited by Croxford L, Frenchman B, Wang J, Qiu Y, Li CH, Song LP, Zhao M
10.4103/1673-5374.175061
How to cite this article: Jia ZQ, Li G, Zhang ZY, Li HT, Wang JQ, Fan ZK, Lv G (2016) Time representation of mitochondrial morphology and function after acute spinal cord injury. Neural Regen Res 11(1)∶137-143.
Funding: This study was supported by the National Natural Science Foundation of China, No. 81272074; the Scientific Research Foundation Project for Doctors in Liaoning Province of China, No. 20121094; Aohongboze Graduate Sci-tech Innovation Foundation, the President Fund of Liaoning Medical University of China, No. 2013003.
http://www.nrronline.org/
Accepted: 2015-12-20
*Correspondence to: Gang Lv, M.D. or Zhong-kai Fan, M.D., ganglv2014jz@163.com or fzkdoctor@163.com.

- 中国神经再生研究(英文版)的其它文章
- Direct reprogramming of somatic cells into neural stem cells or neurons for neurological disorders
- Vascular endothelial growth factor: an attractive target in the treatment of hypoxic/ischemic brain injury
- Glucocorticoids and nervous system plasticity
- RhoA/Rho kinase in spinal cord injury
- The potential of neural transplantation for brain repair and regeneration following traumatic brain injury
- Letter from the Editors-in-Chief