
慢粒灵胶囊药用成分质量控制研究*
2016-12-06 03:04贾勇崔志海赵金秀
西部医学 2016年11期
贾勇 崔志海 赵金秀
(1.廊坊市中医院制剂室, 河北 廊坊 065000;2.廊坊市药检所, 河北 廊坊 065000;3.廊坊市中医院中药房, 河北 廊坊 065000)
·论著·
慢粒灵胶囊药用成分质量控制研究*
贾勇1崔志海2赵金秀3
(1.廊坊市中医院制剂室, 河北 廊坊 065000;2.廊坊市药检所, 河北 廊坊 065000;3.廊坊市中医院中药房, 河北 廊坊 065000)
目的 建立慢粒灵胶囊的青黛、山豆根、红花薄层色谱鉴别及高效液相色谱法测定该制剂中的靛玉红含量。方法 青黛、山豆根、红花采用薄层色谱法(TLC)硅胶G层析板进行定性鉴别;采用高效液相色谱(HPLC)法测定该制剂中的靛玉红含量。色谱柱Inertsil ODS-SP 5 μm 4.6×250 mm,流动相甲醇-水(70∶30),检测波长为292 nm,流速1.0 ml/min,柱温35 ℃;结果 TLC色谱中斑点清晰,易于识别。靛玉红在0.051~0.51 μg范围内呈良好的线性关系(r=0.9999),平均回收率为98.6%,RSD=1.9%(n=6)。结论 青黛、山豆根、红花薄层色谱及高效液相色谱法测定靛玉红含量。该方法简单,准确,专属性好、重现性稳定,灵敏度高,对慢粒灵胶囊制剂的制备与检测提供科学依据。
慢粒灵胶囊; 靛玉红; TLC; HPLC
慢粒灵胶囊为老院长验方,由廊坊市中医院制剂室生产。慢粒灵胶囊由青黛、山豆根、三棱、莪术、红花等药味组成,具有活血化瘀,消癥散结,用于慢性粒细胞白血病治疗。为了有效的控制产品的内在质量,采用高效液相色谱法HPLC对其主要药味青黛中的有效成分靛玉红进行了定量检测,制定了山豆根、红花、青黛的定性鉴别,对该制剂的制备与检测提供科学依据。
1 仪器与试药
1.1 仪器 Shimadzu 2010-AHT 高效液相色谱仪(日本岛津公司,DAD检测器),电子天平(日本岛津Auw120d);KQ2200B型超声波清洗器(功率100 W,频率40 Hz,昆山市超声仪器有限公司)。
1.2 药品与试剂 靛玉红对照品 来源于中国食品药品检定研究院(批号 110717-200204,供含量测定用);苦参碱对照品来源于中国食品药品检定研究院(110805-200508,国家药品标准物质);红花对照药材 中国食品药品检定研究院(120907-201412,国家药品标准物质)。甲醇为HPLC级,水为娃哈哈纯净水,其余试剂均为分析纯试剂。慢粒灵胶囊及缺有相关药味的阴性对照样品均由廊坊市中医院制剂室提供,慢粒灵胶囊暂时没有批号。
2 方法与测定结果
2.1 青黛的鉴别 取慢粒灵胶囊10粒,混匀,称量4 g, 加甲醇溶剂50 ml水浴回流处理30分钟,滤过后,滤液蒸干处理;加30 ml水使溶解,置于分液漏斗中,采用水饱和后的正丁醇溶液萃取2次,每次为30 ml; 合并正丁醇提取溶液,采用100 ℃水浴蒸干,蒸干后用1 ml甲醇溶解,作为样品溶液[1]。另取购自中检院靛玉红对照品1 mg,加甲醇1 ml,混匀,作为对照品溶液。再取处方中不含青黛药材的制剂同法制成阴性对照溶液。参照中国药典2015年版四部 0502试验,吸取上述三种溶液各10 μL,以甲苯-三氯甲烷-丙酮(5∶4∶1)为展开剂,点与薄层层析用的硅胶G板上,薄层展开,取出,晾干。供试品与对照品相应位置显相同颜色斑点,而阴性样品再相应位置未显斑点,阴性对照无干扰。结果见图1。

图1 青黛TLC图
Figure 1 TLC of indigo naturalis
注:1.慢粒灵胶囊(20151101); 2.慢粒灵胶囊(20151120);3.慢粒灵胶囊(20160104); 4.靛玉红对照品;5.阴性供试品
2.2 山豆根的鉴别 取慢粒灵胶囊10粒,置于锥形瓶中,加入甲醇60 ml加热回流30分钟处理,冷却后过滤,滤液水浴蒸干;蒸干后残渣加水40 ml溶解,加盐酸调节PH为3~4,用乙醚50 ml,分两次萃取,弃去乙醚液,分取水层,加10%氢氧化钠溶液调PH值为13,置于分液漏斗中,用氯仿液30 ml提取3次;合并氯仿提取液,蒸干,蒸干后用甲醇1 ml使溶解,制成样品溶液[2]。另取购自中检院苦参碱对照品1 mg,加甲醇1 ml,混匀,作为对照品溶液。作为对照品溶液。再取去除处方中山豆根药材同法制成阴性对照溶液。参照中国药典2015年版四部 0502试验,吸取上述制备的样品溶液、阴性样品溶液分别为10 μL,苦参碱溶液5 μL,以氯仿-甲醇-浓氨试液(4∶1∶0.1)为展开剂,点与薄层层析用的硅胶G板上,薄层展开,取出,晾干。喷以稀碘化铋钾试液[3]。供试品与对照品相应位置显相同颜色斑点,而阴性样品再相应位置未显斑点,阴性对照无干扰。结果见图2。
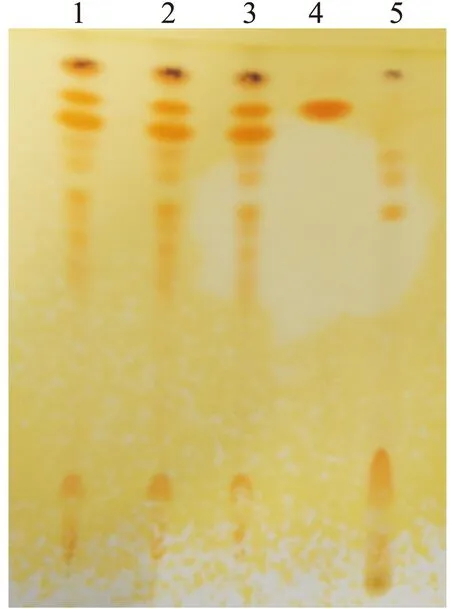
图2 山豆根TLC图
Figure 2 TLC of Radix Sophorae Subprostratae
注:1.慢粒灵胶囊(20151101); 2.慢粒灵胶囊(20151120); 3.慢粒灵胶囊(20160104); 4.苦参碱对照品; 5.阴性供试品
2.3 红花的鉴别 取慢粒灵胶囊10粒, 混匀,称量4 g, 加入50 ml甲醇试剂水浴回流加热30分钟,滤过,滤液蒸干;加30 ml水使溶解,置于分液漏斗中,采用水饱和后的正丁醇溶液萃取2次,每次为30 ml;合并正丁醇提取溶液,采用100 ℃水浴蒸干,蒸干后残渣加1 ml甲醇溶解,作为样品溶液。另取购自中检院红花对照药材1 g,加水200 ml,煎煮1小时,快速滤过,滤液采用减压浓缩至约10 ml,用水饱和的正丁醇萃取2次,每次为30 ml,合并正丁醇提取溶液,采用100 ℃水浴蒸干,蒸干后用1 ml甲醇溶解,作为对照药材溶液,再取去除处方中红花药材,采用同样方法,制成阴性对照溶液。参照中国药典2015年版四部 0502试验,吸取上述三种溶液各5 μL,以乙醚-三氯甲烷(2∶3)为展开剂,点与薄层层析用的硅胶G板上,薄层展开,置于预先用展开剂饱和15分钟处理的展开缸内,展开处理,取出,挥干溶剂,在氨蒸气中熏15分钟,置于365 nm紫外灯下检视[4]。供试品色谱中在阴性对照无干扰,在与对照药材色谱相应的位置,显相同颜色的荧光主斑点,结果见图 3。
2.4 靛玉红的含量测定2.4.1 色谱条件 色谱柱Inertsil ODS-SP 5 μm 4.6×250 mm C/N.5020-01732;流动相 甲醇-水(70∶30);流速1.0 ml/min,柱温35 ℃;检测波长为292 nm[5]。

图3 红花TLC图
Figure 3 TLC of carthamus tinctorious
注:1.慢粒灵胶囊(20151101); 2.慢粒灵胶囊(20151120); 3.慢粒灵胶囊(20160104);4.红花对照药材; 5.阴性供试品
2.4.2 对照品溶液制备 精密称取国家药品标准物质的靛玉红对照品12.68 mg,置250 ml量瓶中, 加三氯甲烷-甲醇(1∶9)稀释至刻度,混匀后,即得(制成1 ml含50.72 μg的对照品溶液)。
2.4.3 供试品溶液制备 取慢粒灵胶囊20粒内容物,研细,精密称取2 g,置锥形瓶中,加入乙醚40 ml,超声处理30分钟,用乙醚洗涤滤渣和滤器;用10%氢氧化钠10 ml洗涤,分取乙醚层,挥干,残渣加三氯甲烷-甲醇(1∶9)溶解,定溶于10 ml量瓶中,采用三氯甲烷-甲醇(1∶9)稀释至刻度,摇匀,0.45 μm微孔滤膜滤过即得[6]。
2.4.4 阴性对照样品溶液的制备 取去除青黛药材处方,制成阴性对照的制剂样品。按照“2.4.3供试品溶液制备”方法制备成待测的阴性样品溶液[7]。
2.4.5 加样回收率试验结果见图4。

图4 靛玉红空白图谱、靛玉红图谱与阴性供试品(C)的HPLC图谱
2.4.6 专属性检测,注入HPLC仪器中阴性对照品溶液、对照品溶液与供试品溶液各10 μL,供试品溶液呈现出于靛玉红对照品溶液相同的色谱峰,阴性样品未出现相同的色谱图形,结果表示阴性对照样品无干扰[8],见图4。
2.4.7 线性范围 精密量取2.4.2项下对照品溶液1、2、4、6、8、10 μL,按照“2.4.1色谱条件”注入液相色谱仪,记录色谱图,以靛玉红浓度为横坐标,靛玉红峰面积为纵坐标,进行线性回归方程,得线性回归方程 Y=108484.1x+7906.495,r=0.9999 得到结果,靛玉红在0.051~0.51 μg,呈现良好的线性关系[9-10]。
2.4.8 精密度检测 取靛玉红对照品溶液浓度为50.72 μg/mL的溶液,采用上述“2.4.1”色谱条件,进样量为10 μL,6次连续进样,采用6次测定峰面积值[11-13],计算RSD为 0.14%。
2.4.9 重复性试验 采用批号为20151120供试品6份, 进样量为10 μL,注入HPLC仪器中,计算6份样品的靛玉红含量为0.209 mg/g,6份样品的RSD值为1.63%。
2.4.10 稳定性试验 取同一供试品溶液(批号20151120),按“2.4.1”色谱条件进行3小时测定一次,共测定6次,进样量为10 μL。6次得到靛玉红峰面积RSD为1.0%,结果说明测定的靛玉红在15小时内基本稳定[14-17]。
2.4.11 回收率测定 取已测定含有量的样品(批号20151120,靛玉红的含量为0.209 mg/g),取约0.5 g,共计6份,精密称定重量,分别置与锥形瓶中,每个锥形瓶中精密加入浓度为0.01024 mg/mL靛玉红对照品溶液10 ml,按2.4.3项下方法制备供试品溶液,测定靛玉红含量,进行回收率计算[18-19],见表1。

表1 靛玉红回收率试验
2.4.12 样品测定 取批号为20151101、20151120、20160104的样品,进行检测,进样量为10 μL。测得含量值[20],结果见图2、表2。

表2 样品测定结果
3 讨论
3.1 青黛鉴别 溶剂考察中采用甲醇、乙醇分别提取、超声处理10分钟,水浴回流10、20、30分钟,结果采用甲醇提取,加热回流30分钟效果好。采用不经过水饱和后的正丁醇、与经过水饱和后的正丁醇溶液萃取比对,结果经过萃取后的供试品斑点清晰干扰少。展开剂的选择,采用甲苯-三氯甲烷(5∶4);三氯甲烷-丙酮(4∶0.5);甲苯-三氯甲烷-丙酮(5∶4∶1),结果甲苯-三氯甲烷-丙酮(5∶4∶1)展开系统好,比移值适中。
3.2 山豆根鉴别 溶剂选择时采用乙醚两次萃取,弃去乙醚液,分取水层,再用氯仿液30ml提取效果好,优于甲醇直接回流后进行薄层分析。采用分别两次PH调节后,杂质干扰少,斑点清晰。展开剂的选择时采用氯仿-甲醇(4∶1);乙酸乙酯-甲醇(4∶1);氯仿-甲醇-浓氨试液(4∶1∶0.1)展开系统好,比移值适中。
3.3 红花鉴别 采用显色时置于预先用展开剂饱和15分钟处理的展开缸内,展开处理,取出,挥干溶剂,在氨蒸气中熏15分钟后,置于365 nm、254 nm紫外灯下与日光下对比检视,结果置于365 nm紫外灯下斑点清晰。
3.4 靛玉红含量测定 通过实验,靛玉红含量测定中,样品用乙酸乙酯水浴,乙醚超声,乙酸乙酯超声,结果证明乙醚超声提取率最高,另外还进行了超声时间条件摸索,20 min,30 min,40 min,结果证明采用30 min提取最佳。靛玉红含量测定色谱条件的优化选择,按《中国药典2015版一部》199页,青黛中靛玉红含量测定色谱条件,波长选择为292 nm。流动相曾选择甲醇∶水=60∶40、甲醇∶水=70∶30、甲醇∶水=80∶20,按照该色谱条件进行测定,结果甲醇∶水=70∶30,分离效果好,靛玉红分离度大于1.5、理论板数高。在保证该方法专属性的同时,大大缩短了样品测定时间,节省了流动相溶剂的使用。
含量测定中曾选择青黛中另一主成分靛蓝的测定。按《中国药典2015版一部》199页,青黛中靛蓝含量测定色谱条件,采用色谱条件为十八烷基硅烷键合硅胶色谱柱,以甲醇-水(75∶25)为流动相,检测波长为606 nm。对照品溶液的制备:取靛蓝对照品2.5 mg,精密称定,置250 ml量瓶中,加2%水合氯醛的三氯甲烷溶液(取水合氯醛,置硅胶干燥器中放置24小时,称取2.0 g,加三氯甲烷至100 ml,放置,出现浑浊,以无水硫酸钠脱水,滤过,即得)约220 ml,超声处理(功率250 W,频率33 kHz)1.5小时,放冷,加2%水合氯醛的三氯甲烷溶液至刻度,摇匀,即得(每1 ml中含靛蓝10 μg)。供试品溶液的制备 取慢粒灵胶囊20粒内容物,研细,精密称取1 g,精密称定,置250 ml量瓶中,加2%水合氯醛的三氯甲烷溶液约220 ml,超声处理(功率250 W,频率33 kHz)30分钟,放冷,加2%水合氯醛的三氯甲烷溶液至刻度,摇匀,滤过,取续滤液,即得。测定法:分别精密吸取对照品溶液与供试品溶液各10 μL,注入液相色谱仪,测定即得。结果分离效果不好,样品中靛蓝理论板数不足500,色谱峰形不对称,分离度小于1.5,故未列入正文。
4 结论
本研究对慢粒灵胶囊的青黛、山豆根、红花薄层色谱鉴别及高效液相色谱法测定靛玉红含量的方法简单,准确,专属性好、重现性稳定,灵敏度高,为慢粒灵胶囊制剂的制备与检测提供了科学依据。
[1]张晓南, 毛云宏,游燕,等.小儿清咽颗粒中青黛的薄层色谱鉴别研究[J].中国民族民间医药,2015,24(11):21-22.
[2]孙旭, 高媛,吴洋,等.复方山豆根合剂质量标准的研究[J].解放军药学学报,2015,31(6):513-515.
[3]丁佩兰, 蒋司嘉,乔春峰,等.苦参碱和氧化苦参碱的分离纯化以及山豆根药材的薄层色谱鉴别[J].中国药学杂志,2004,39(5):333-335.
[4]中国药典[S].一部.2015:151.
[5]中国药典[S].一部.2015:199.
[6]徐斐, 胡安青.HPLC法测定复方青黛胶囊中靛玉红的含量[J].齐鲁药事,2012,31(3):141-142.
[7]马莉, 孙琴,李友,肖小河.HPLC法测定板蓝根药材及制剂中靛蓝和靛玉红含量[J].药物分析杂志, 2010,30(9);1642-1645.
[8]王敏, 马玉梅.安肾丸的质量控制研究[J].现代药物与临床,2012,27(6):571-574.
[9]林雪霞. RP-HPLC法同时测定升麻黄芩颗粒中黄芩苷、芍药苷和葛根素[J]. 中国药师,2015,18(3):485-487.
[10] 周军, 张蕾,王杰. HPLC-DAD法测定脑血栓片中苦杏仁苷、羟基红花黄色素A、芍药苷、阿魏酸、丹酚酸B和丹参酮ⅡA[J].现代药物与临床, 2015,30(3): 262-266.
[11] Radojkovic B, Milic J, Calija B. Compounding of slow-release niacinamide capsules: feasibility and characterization[J]. Int J Pharm Compd,2012,16(5):434-437.
[12] Zur E. Compounding slow-release capsules: a comprehensive review and an Excel spreadsheet for faster calculations of excipients[J]. Int J Pharm Compd,2013,17(1):10-22.
[13] Glowiak DL, Green JL, Bowman BJ. In vitro evaluation of extemporaneously compounded slow-release capsules containing morphine sulfate or oxycodone hydrochloride[J]. Int J Pharm Compd,2005,9(2):157-164.
[14] Bogner RH, Szweijkowski J, Houston A. Release of morphine sulfate from compounded slow-release capsules: the effect of formulation on release[J]. Int J Pharm Compd,2001,5(5):401-405.
[15] Reis EA, Rocha-Leao MH, Leite SG. Slow-release nutrient capsules for microorganism stimulation in oil remediation[J]. Appl Biochem Biotechnol,2013,169(4):1241-1249.
[16] Reis EA, Rocha-Leao MH, Leite SG. Slow-release nutrient capsules for microorganism stimulation in oil remediation[J]. Appl Biochem Biotechnol,2013,169(4):1241-1249.
[17] Webster KD, Al-Achi A, Greenwood R. In vitro studies on the release of morphine sulfate from compounded slow-release morphine-sulfate capsules[J]. Int J Pharm Compd,1999,3(5):409-411.
[18] Rhodes AP. Efficacy of slow-release albendazole capsules in controlling lungworms and gastro-intestinal nematodes in red deer (Cervus elaphus)[J]. N Z Vet J,1993,41(3):131-133.
[19] Doser K, Guserle R, Nitsche V,etal. Comparative steady state study with 2 fenofibrate 250 mg slow release capsules. An example of bioequivalence assessment with a highly variable drug[J]. Int J Clin Pharmacol Ther,1996,34(8):345-348.
[20] Heusinger JH, Breuel HP, Behrendt WA,etal. Pharmacokinetics of theophylline. Repeated single evening administration of Pulmo-Timelets slow-release capsules in comparison with theophylline slow-release tablets[J]. Fortschr Med,1989,107(32):692-696.
Control of quality standards for Manliling Capsule
JIA Yong1, CUI Zhihai2, ZhAO Jinxiu3
(1.DispensingRoom,LangfangTCMHospital,Langfang065000,Hebei,China;2.LangfangInstituteforDrugControl,Langfang065000,Hebei,China;3.TCMPharmacy,LangfangTCMHospital,Langfang065000,Hebei,China;)
Objective To establish the quality standard for Manliling Capsule. Methods Naturalis Sophorae Tonkinensis Radix et Rhizoma and Carthami Flos were identified by TLC. Indirubin was determined by HPLC. Indigo Naturalis Sophorae Tonkinensis Radix et Rhizoma and Carthami Flos were identified by thin-layer chromatography (TLC) silica gel G.. Indirubin was determined by HPLC. The sample was separated by Inertsil ODS-SP 5μm 4.6×250 mm with methanol -water (70:30)in gradient elution. The detection wavelength was 292 nm. The flow rate was 1.0 ml/min, and the column temperature was 35℃. Results Indigo Naturalis, Sophorae Tonkinensis Radix et Rhizoma and Carthami Flos could be detected by TLC. Indirubin showed a good linear relationship in a range of 0.051~0.51μg and the average recovery was 98.6%, RSD was 1.9%n=6). Conclusion The quality standard for Manliling Capsule. Naturalis Sophorae Tonkinensis Radix et Rhizoma and Carthami Flos were identified by TLC. Indirubin was determined by HPLC. The methods are simple, sensitive and accurate with good reproducibility, methods may be used for the quality control of Manlilng Capsule.
Manliling Capsule; Indirubin; TLC; HPLC
HPLC of Indirubin
ubstance (A), sample (B)and negative sample(C)
河北省科技厅科技攻关项目(2013276163)
R 914
A
10.3969/j.issn.1672-3511.2016.11.029
2016-05-23; 编辑: 张文秀)
猜你喜欢
化工管理(2022年14期)2022-12-02
中国化肥信息(2022年3期)2022-05-05
食品安全导刊(2021年36期)2021-03-14
科学导报(2020年75期)2020-12-21
山西中医药大学学报(2020年2期)2020-06-06
石油石化绿色低碳(2019年6期)2019-02-13
中成药(2018年12期)2018-12-29
中国化肥信息(2016年27期)2016-05-17
Asian Journal of Urology(2015年3期)2015-12-16
治淮(2013年1期)2013-09-10
