
贮存期间硝化甘油的分解机理
2017-05-07 01:26裴立冠董可海唐岩辉李文佐
含能材料 2017年10期
裴立冠, 董可海, 唐岩辉, 张 波, 于 畅, 李文佐
(1. 海军航空工程学院飞行器工程系, 山东 烟台 264001; 2. 烟台大学化学化工学院, 山东 烟台 264005)
1 引 言
硝化甘油(三硝酸甘油酯,NG)作为硝酸酯增塑聚醚推进剂(NEPE)及双基推进剂的一种重要增塑剂[1],在实际贮存中受热影响会进行包括NO2基团消去和HONO消除两种反应通道在内的分解,释放出NO2等气体(自分解反应)[2],同时受生成的NO2的催化作用又会产生α位和β位夺氢两种反应通道在内的自催化反应[3]。上述四条反应通道将最终导致推进剂长期贮存期间硝化甘油含量减少,降低推进剂安定性并加速其老化[4-5],因此理清硝化甘油分解机理,对研究推进剂老化规律具有重要意义。
关于硝化甘油分解的研究开始于20世纪70年代。张林军等[4]通过液相色谱法获取了改性双基推进剂中硝化甘油在热老化和湿热老化条件下的分解情况及其对推进剂硬度和燃速的影响; 丁黎等[5]研究了NEPE推进剂各组分对硝化甘油的分解过程的作用; 姚子云等[6]采用高压差热分析仪和傅里叶变换原位红外光谱仪得出自催化动力方程可用于描述硝化甘油在压力下的分解情况; Roos等[7]和Hiyoshi等[8]采用T-Jump/FTIR热解实验得到硝化甘油在不同温度和压力下CO2、CO、NO2等共性分解产物; Chin A等[9]通过电子自旋共振谱技术提出O—NO2的均裂是硝化甘油分解反应的引发点; Musanic等[10]采用差示扫描量热法发现加热速度和样品量与双基推进剂中硝化甘油的分解具有直接关系。另外,还有少部分研究者采用量子力学方法从理论上探讨硝化甘油反应机理,如肖鹤鸣等[11]采用该方法计算了硝化甘油热裂解和β—H转移消除反应的位能曲线,并得出O—NO2为分子中最弱键; 颜庆丽等[2]运用密度泛函理论详细分析了硝化甘油自分解反应中的NO2基团消去和HONO消除两种反应通道的分解机理; 裴海潮等[3]在文献[6]的基础上,运用量子力学方法研究了α位和β位夺氢两种自催化反应机理。
尽管目前对于硝化甘油最终分解产物及过程中重要的反应节点已经有较为清晰的认识,但实验所采用的方法大多数为高温热解,且尚未有针对其分解各阶段主要反应通道的报道,对于硝化甘油在贮存期间分解反应类型随时间的变化情况还不清楚。硝化甘油易于分解,且超过70 ℃就有爆炸危险[12],而其在常温下反应缓慢,不利于浓度监测,为此,本研究通过热分解实验(55 ℃和60 ℃)对硝化甘油贮存期间分解过程中三种气体(CO、NO、NO2)浓度进行测量,并采用密度泛函理论的B3LYP方法及变分过渡态理论计算了四种反应通道初始反应的速率常数,以期从定性和定量两方面研究硝化甘油在贮存期间的分解反应,得到反应各个阶段的可能模式。
2 热分解实验与分析
2.1 样品及仪器设备
(1) 硝化甘油样品250 g,航天科工41所制备,纯度达99.62%。
(2) 高低温试验箱,无锡腾发设备有限公司生产; 复合式气体检测仪(CO、NO、NO2,量程0~10 mg·kg-1,分辨率0.01 mg·kg-1),华瑞科力恒有限公司生产; 密封袋(1000 mL)。
2.2 实验条件与方法
通过转接接头把传感器与密封袋连接起来,形成密闭空间。将硝化甘油样品放于密封袋后,分别置于55 ℃和60 ℃的高低温试验箱中,定时取样监测硝化甘油产生的特定气体浓度(复合式气体检测仪的泵流量调节至0.8 L·min-1),每种温度下试验样品为6组,监测结果取平均值。
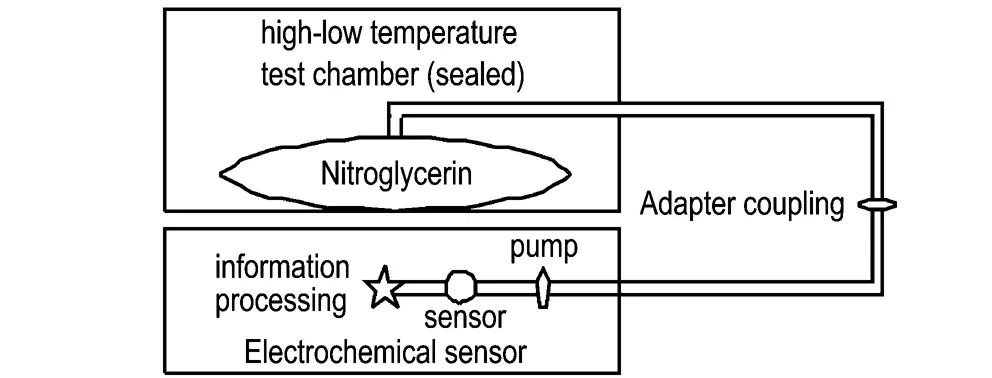
图1实验装置示意图
Fig.1Schematic diagram of the experimental device
2.3 结果与讨论
基于热分解实验,最终得到了CO、NO、NO2浓度随时间的变化情况(图2),硝化甘油在60 ℃时的分解速度为0.28 μg·(g·h)-1[12],由此可知本实验中样品量充足,气体浓度变化基本不会受硝化甘油样品量减少的影响。对于三种气体,据硝化甘油四种反应通道分解机理可知[2-3],NO2主要源于α位和β位O—NO2键均裂,即NO2基团消去反应,NO主要由HONO消除、α位夺氢和β位夺氢三种反应通道生成的HONO进一步分解产生,而CO为四种反应通道的共性气体。在此,根据图2中气体浓度变化情况,对硝化甘油分解过程进行定性分析。

a. 55 ℃
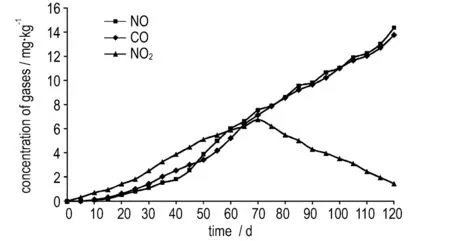
b. 60 ℃
图255 ℃和60 ℃下气体浓度变化
Fig.2Changes in concentrations of gases at 55 ℃ and 60 ℃
(1) NO2气体浓度在两种温度下较CO、NO优先测得,证明NO2基团消去反应为贮存期间硝化甘油分解的引发点。该气体浓度首先随分解时间的增长而逐渐增大,当到达节点时间时(55 ℃下为90天,60 ℃下为70天),浓度达到最大值,然后随着分解时间的增长NO2浓度逐渐减小,证明此时NO2生成速率小于NO2消耗速率。研究表明,硝化甘油分解过程中,NO2是重要的中间产物,NO2基团消去反应中O—NO2的均裂会产生大量的NO2气体,而该NO2气体又会进一步催化硝化甘油分解产生自催化反应的α位和β位夺氢[13]。由此可以得出,小于节点时间时,受制于NO2产气量的限制,自分解反应中的NO2基团消去为硝化甘油分解重要的反应通道,当大于节点时间时,自催化反应为主要反应形式。
(2)NO和CO两种气体浓度随着热分解时间的增长逐渐增大,但在初始阶段,两种气体浓度小于NO2浓度,证明在该阶段其他三种反应通道速率均小于自分解反应中的NO2基团消去反应。随着分解反应的进行,在节点时间后,两种气体的浓度均超过NO2浓度,且持续增大,证明此时HONO消除、α位夺氢和β位夺氢三种反应通道中至少有一种通道反应速率较大。
综上,本研究以节点时间为基准,将硝化甘油贮存期间的分解过程划分为Ⅰ、Ⅱ两个阶段,反应Ⅰ阶段自分解反应中的NO2基团消去反应为重要的反应通道,反应Ⅱ阶段中自催化反应为重要反应形式。
3 量子化学计算与分析
通过硝化甘油热分解实验最终得到了硝化甘油分解反应随时间变化的定性分析结果,但依然无法对每个阶段内部不同反应通道做出定量比较,而密度泛函理论(Density functional theory,DFT)正是目前研究此类化学反应问题的一种重要量子力学分析方法[13]。为此,本研究采用DFT/B3LYP方法,在6-31G**基组水平上研究各个反应通道的起始反应,并采用变分过渡态理论计算化学反应速率常数,以确定硝化甘油详细反应机理。
3.1 反应机理研究
3.1.1 计算方法
硝化甘油分解反应的四种反应通道反应复杂,过程中包含众多中间体[2-3]。本研究以四种反应通道的起始反应(式(1)、(2)、(3)、(4))为研究对象,运用B3LYP/6-31G**方法,对反应物、过渡态(TS1、TS2、TS3、TS4)、产物进行优化,计算单点能和振动频率,并通过频率分析和内秉坐标计算(IRC)确认结构的合理性,上述工作均在Gaussian09程序包完成[14]。
Path1:
(a) CH2ONO2CHONO2CH2ONO2(R)→
CH2ONO2CHONO2CH2O·(Ⅰ1)+·NO2
(b) CH2ONO2CHONO2CH2O·(Ⅰ1)→TS1→
CH2ONO2CHOHCHO(P1)+·NO2
(1)
Path2:
CH2ONO2CHONO2CH2ONO2(R)→TS2→
CH2ONO2CHONO2CHO(P2)+HONO
(2)
Path3:
CH2ONO2CHONO2CH2ONO2(R)+NO2→TS3→CH2ONO2CHONO2CHONO2(P3)+HONO
(3)
Path4:
CH2ONO2CHONO2CH2ONO2(R)+NO2→TS4→CH2ONO2CONO2CH2ONO2(P4)+HONO
(4)
Path1为NO2基团消去反应通道,(a)表示α位O—NO2键直接均裂,目前还尚未发现过渡态[2,15],(b)表示β位O—NO2键均裂,由于(a)反应为直接断裂,因此从过渡态理论角度,此通道(b)为实际起始反应,也是后续速率常数的计算对象。Path2~Path4分别表示HONO消除、α位夺氢和β位夺氢三种反应通道。
3.1.2 几何优化结果
优化后四个起始反应中反应物、过渡态及产物的几何结构(图3),键长与文献[2]、[3]、[11]中优化结果差值为0.01~0.03 Å,可见优化结果有效,其中TS1、TS2、TS3、TS4四种过渡态对应的唯一虚频分别是1881i,1489i,1569i,1765i cm-1。

图3反应物、中间体和产物的优化结构图
Fig.3Optimized structures for reactants, intermediates and products
3.1.3 反应路径讨论
图4列出了四种起始反应的势能变化情况(所有能量均为零点振动能(ZPE)校正后的能量)。由图4可知,四种起始反应生成物能量都低于反应物能量,即均为放热反应。对于Path1,首先吸热进行α位O—NO2键的直接均裂,所需键解离能为117.13 kJ·mol-1,形成中间体Ⅰ1。然后Ⅰ1经TS1最终生成P1,此过程中Ⅰ1分子上一个α位C—H键长由1.09 Å增长为TS1对应的1.36 Å,所需键离解能为120.56 kJ·mol-1。Path2所需活化能为122.40 kJ·mol-1,过程中α位O—N键长从1.45 Å增长到2.03 Å,而此位置上一个C—H键长由1.09 Å增长为1.31 Å。自催化反应的两条反应通道Path3和Path4所需活化能均小于自分解反应通道,分别为100.89 kJ·mol-1和90.46 kJ·mol-1。其中,对于Path3放热反应过程,α位上一个C—H键长由1.09 Å增长到1.41 Å,与此同时该H原子与NO2分子上一个O原子距离从3.42 Å缩短为1.18 Å。Path4中,β位上一个C—H键长从1.09 Å增长为1.41 Å,而该H原子与NO2分子上一个O原子距离由2.91 Å缩短为1.20 Å。

图4四种起始反应势能变化图
Fig.4Change in potential energy for four kinds of initial reactions
3.2 反应分子动力学研究
3.2.1 计算方法
在硝化甘油分解反应的四种反应通道中,各反应通道的起始反应在各反应全过程中所需活化能最大,均为主要的反应速率控制步骤,故其速率常数能够代表整个反应通道的反应快慢[16]。而小曲率隧道效应校正的变分过渡态理论(Canonical variational transition state theory combined with a small-curvature tunneling correction, CVT/SCT)对多种类型化学反应速率常数的分析表明该方法计算值与实验值相近,是一种有效的速率常数计算方法[17-18]。为此,本研究采用CVT/SCT方法,对四种反应通道的化学反应速率常数进行计算,此步骤在Polyrate 9.7软件包下[19]进行。硝化甘油在温度低于15 ℃时容易结晶且相互间摩擦可能产生爆炸危险,因此将20~60 ℃作为所要研究的贮存温度区间。
(5)
式中,kCVT/SCT(T)表示采用CVT/SCT方法得到的化学反应速率常数;σ为对称因子;kB为Boltzmann常数,J/K;T为反应温度,K;h为普朗克常数,J·s;s为反应坐标;QGT(T,s)为温度T时,反应坐标s点的经典配分函数;ΦR(T)为单位体积内反应物配分函数;VMEP(s)为QGT(T,s)的能量零点;μSCT为小曲率隧道效应校正系数。
3.2.2 结果与讨论
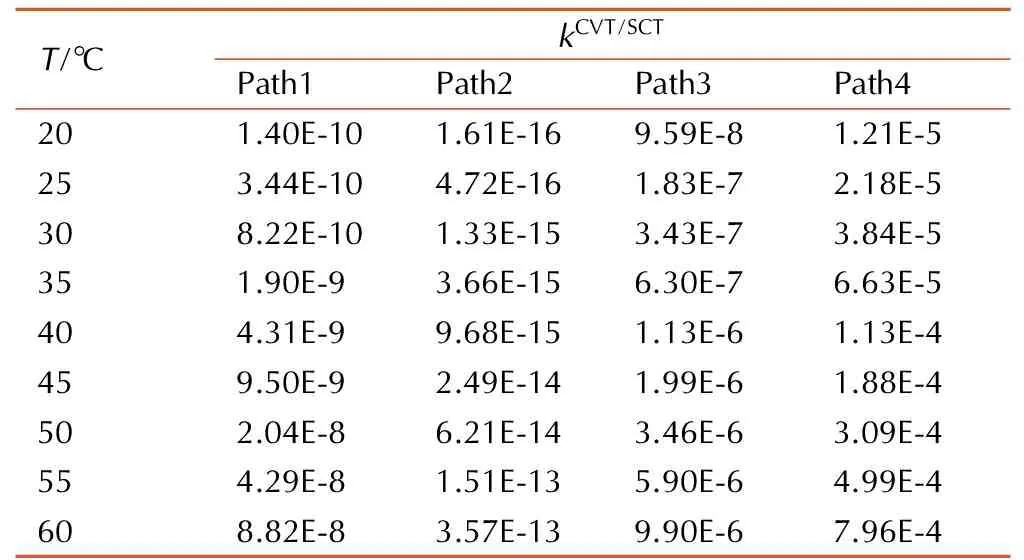
基于密度泛函理论分析结果,采用CVT/SCT方法计算得到四种起始反应在20~60 ℃的化学反应速率常数,结果见表1。
表120~60 ℃下四种起始反应速率常数
Table1The rate constants of four kinds of initial reactions at 20-60 ℃
T/℃kCVT/SCTPath1Path2Path3Path4201.40E⁃101.61E⁃169.59E⁃81.21E⁃5253.44E⁃104.72E⁃161.83E⁃72.18E⁃5308.22E⁃101.33E⁃153.43E⁃73.84E⁃5351.90E⁃93.66E⁃156.30E⁃76.63E⁃5404.31E⁃99.68E⁃151.13E⁃61.13E⁃4459.50E⁃92.49E⁃141.99E⁃61.88E⁃4502.04E⁃86.21E⁃143.46E⁃63.09E⁃4554.29E⁃81.51E⁃135.90E⁃64.99E⁃4608.82E⁃83.57E⁃139.90E⁃67.96E⁃4
Note:Tis the reaction temperature;kCVT/SCTis the calculated rate constant by the canonical variational transition state theory combined with a small-curvature tunneling correction.
由表1可知,在20~60 ℃,四种起始反应中,Path2速率最慢,为其他三种反应速率的1/1011~1/105。Path4速率最快,为其他三类反应速率的102~1011倍。而Path3反应速率较Path1快约两个数量级。基于表1中的数据,拟合出了四种起始反应在所研究温度区间内的速率常数三参数表达式:
(6)
(7)
(8)
(9)
综上所述,可将反应Ⅰ阶段和反应Ⅱ阶段作进一步分析,反应Ⅰ阶段由于NO2气体产生量的限制,主要进行硝化甘油自分解反应,其中NO2基团消去反应为主反应通道,HONO消除反应以较低速率进行,并且此时自催化反应随着自分解反应产生的NO2浓度逐渐增大反应速率逐渐加快。当NO2气体达到一定浓度后进入反应Ⅱ阶段,β位夺氢自催化反应成为主反应通道,α位夺氢自催化反应为次反应通道,NO2基团消去反应和HONO消除反应随贮存时间的增长对硝化甘油分解的贡献率持续降低。
4 结 论
(1) 基于热分解监测实验可知,在55 ℃和60 ℃条件下,硝化甘油热分解释放出的NO和CO两种气体浓度随着分解时间的增长逐渐增大。而NO2气体先于其它两种气体产生,证明NO2基团消去反应为贮存期间硝化甘油分解的引发点,且该气体浓度变化呈现先增大后减小的规律。
(2) 在20~60 ℃内硝化甘油分解反应的四种反应通道中,β位夺氢自催化反应速率最快,为其他三类反应速率的102~1011倍,HONO消除自分解反应速率最慢,为其他三种反应速率的1/1011~1/105,而α位夺氢反应速率较NO2基团消去反应快约两个数量级。
(3) 综合理论与实验结果,得出硝化甘油贮存期间的分解反应可以NO2气体达到最大浓度值的时间点为基准划分为两个阶段,反应Ⅰ阶段NO2基团消去反应为主反应通道,反应Ⅱ阶段β位夺氢自催化反应为主反应通道,α位夺氢自催化反应通道为次反应通道,而HONO消除自分解反应在整个反应过程中对硝化甘油分解的贡献率最小。
参考文献:
[1]庞爱民,马新刚,唐承志. 固体火箭推进剂理论与工程[M]. 北京: 中国宇航出版社, 2014.
PANG Ai-min, MA Xin-gang, TANG Cheng-zhi. Solid rocket propellant theory and engineering [M]. Beijing: China Astronautic Publishing House, 2014.
[2]YAN Qing-li, ZHU Wei-hua, PANG Ai-min, et al. Theoretical study on the unimolecular decomposition of nitroglycerin [J].JournalofMolecularModeling, 2013, 19(4): 1617-1626.
[3]裴海潮,吴婉娥,付潇,等.二氧化氮催化硝化甘油分解机理的数值计算[J].固体火箭技术,2016, 39(4): 538-541.
PEI Hai-chao, WU Wan-e, FU Xiao, et al. Numerical simulation on catalytic decomposition mechanism of NO2on nitroglycerin [J].JournalofSolidRocketTechnology, 2016, 39(4): 538-541.
[4]张林军,张冬梅,王琼,等.湿热环境对浇铸改性双基推进剂力学和燃烧性能的影响[J].化学推进剂与高分子材料,2013,11(2): 77-79.
ZHANG Lin-jun, ZHANG Dong-mei, WANG Qiong, et al. Influence of hydrothermal environment on mechanical and combustion performance of cast modified double base propellant [J].ChemicalPropellants&PolymericMaterials, 2013,11(2): 77-79.
[5]丁黎,赵风起,潘清,等.原位傅里叶变换红外光谱研究含CL-20的NEPE推进剂的热分解[J]. 火炸药学报,2008,31(4): 77-81.
DING Li, ZHAO Feng-qi, PAN Qing, et al. Research on the thermal decomposition of NEPE propellant containing CL-20 by Situ/RSFT-IR [J].ChineseJournalofExplosives&Propellants, 2008, 31(4): 77-81.
[6]姚子云,罗秉和.硝化甘油在压力下的热分解动力学及机理[J].中北大学学报,1989,10(2): 42-48.
YAO Zi-yun, LUO Bin-he. Thermal decomposition dynamics and mechanism of nitroglycerin under pressure [J].JournalofNorthUniversityofChina, 1989, 10(2): 42-48.
[7]ROOS B D, BRILL T B. Thermal decomposition of energetic materials 82 correlations of gaseous products with the composition of aliphatic nitrate esters [J].CombustingandFlame, 2002, 128(1-2): 181-190.
[8]HIYOSHI R I, BRILL T B. Thermal decomposition of energetic materials 83 comparison of the pyrolysis of energetic materials in air versus argon [J].Propellants,Explosives,Pyrotechnics, 2002, 27(1): 23-30.
[9]CHIN A, ELLISON D S, POEHLEIN S K. Investigation of the decomposition mechanism and thermal stability of nitrocellulose/nitroglycerine based propellants by election spin resonance [J].Propellants,Explosives,Pyrotechnics, 2007, 32(2): 117-126.
[10]MUSANIC S M, HOURA I F, SUCESKA M. Applicability of non-isothermal DSC and Ozawa method for studying kinetics of double base propellant decomposition [J].CentralEuropeanJournalofEnergeticMaterial,2010,7(7): 233-251.
[11]肖鹤鸣,王大喜.硝酸酯类的量子化学研究—硝化甘油的热解原理[J].兵工学报,1992,2(1):41-46.
XIAO He-ming, WANG Da-xi. Quantum chemical studies on the nitrate esters-trinitrogl ycerin and mechanisms of its thermal decomposition[J].ActaArmamentarii, 1992,2(1): 41-46.
[12]黎留鑫.硝化甘油安全生产技术[J].火炸药学报,1984,3,7(1): 27-35.
LI Liu-xin. Production safety technology of nitroglycerin[J].ChineseJournalofExplosives&Propellants, 1984,3,7(1): 27-35.
[13]R Wang, D Zhang, C Liu. DFT study of the adsorption of 2,3,7,8-tetrachlorodibenzo-p-dioxin on pristine and Ni-doped boron nitride nanotubes[J].Chemosphere, 2017, 168(2): 18-24.
[14]FRISCH M J, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 09,Revision A. 1[CP],Gaussian Inc. ,Wallingford CT, 2009.
[15]Gong C D, Xiao H M. Studies on the molecular structures, vibrational spectra and thermodynamic properties of organic nitrates using density functional theory and ab initio methods [J].JournalofMolecularModeling, 2001, 572: 213-221.
[16]JOSEPH R Murdoch. What is the rate-limiting step of a multistep reaction? [J].JournalofChemicalEducation,1981, 58: 32-36.
[17]霍瑞萍,张祥,黄旭日,等. C4H(2∑+)+H2反应机理的直接动力学研究[J].化学学报,2013,71(5): 743-748.
HUO Rui-ping, ZHANG Xiang, HUANG Xu-ri, et al. Mechanism study of C4H(2∑+)+H2reaction by direct Ab initio methods [J].ActaChimicaSinica, 2013,71(5): 743-748.
[18]张田雷,杨晨,凤旭凯,等.HS+HO2气相反应机理及主通道速率常数的理论研究[J]. 物理化学学报,2016, 32 (3): 701-710.
ZHANG Tian-Lei, YANG Chen, FENG Xu-kai, et al. Theoretical study on the atmospheric reaction of HS with HO2: mechanism and rate constants of the major channel [J].ActaPhysico-ChimicaSinica, 2016, 32 (3): 701-710.
[19]CORCHADO J C, CHUANG Y Y, FAST P L, et al. POLYRATE version 9.7 [CP], University of Minnesota, Minneapolis, MN, 2007.
猜你喜欢
含能材料(2022年8期)2022-08-13
数学年刊A辑(中文版)(2021年1期)2021-06-09
铜仁学院学报(2018年6期)2018-07-05
新高考·高一物理(2016年3期)2016-05-18
浙江理工大学学报(自然科学版)(2015年5期)2015-03-01
西安建筑科技大学学报(自然科学版)(2014年5期)2014-11-10
火炸药学报(2014年5期)2014-03-20
火炸药学报(2014年5期)2014-03-20
火炸药学报(2014年1期)2014-03-20
郑州大学学报(理学版)(2013年3期)2013-03-11
