
Notch1 activation protects against triptolide-induced oxidative damage and apoptosis in hepatocytes*
2017-06-19 15:59,,,,,,
中山大学学报(自然科学版)(中英文) 2017年3期
, , , , , ,
(School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China)
Notch1 activation protects against triptolide-induced oxidative damage and apoptosis in hepatocytes*
XIONGZhewen,WANGLi,KONGJiamin,WANGWenwen,CHENGYisen,SHENFeihai,HUANGZhiying
(School of Pharmaceutical Sciences, Sun Yat-sen University, Guangzhou 510006, China)
Triptolide (TP), the predominant active component of the traditional Chinese herbTripterygiumwilfordiiHook f. (TWHF), has been shown to exert multiple pharmacological effects. However, the clinical applications of TP are limited by its severe adverse effects, especially hepatotoxicity. The aim of this study was to investigate the potential protective effects of Notch1/NF-E2-related factor 2 (Nrf2) signaling on TP-induced hepatotoxicity. In the present study, TP caused oxidative stress and cellular apoptosis in Chang liver cells, which were accompanied by inhibition of the Notch1 activation signal. In addition, the activation of Notch1 by Jagged1 protected against TP-induced cytotoxicity, whereas the inhibition of Notch1 by DAPT or Notch1 siRNA increased the damage. We also found that activation of Notch1 upregulated Nrf2 and heme oxygenase-1 (HO-1) expression and that the inhibition of Notch1 resulted in the opposite effect. Interestingly, a reduction in Nrf2 almost eliminated the protective effect conferred by Jagged1, which indicated that Nrf2 was essential in the protection against TP-induced hepatotoxicity through the activation of Notch1. Overall, these results suggested that Notch1 signaling ameliorated the detrimental effects of TP by decreasing oxidative stress and downregulating apoptosis, and that the Nrf2 signaling pathway was a key downstream mediator of this effect.
triptolide; Notch1; Nrf2; oxidative stress; apoptosis
Triptolide (TP) is a diterpenoid epoxide, originally purified from the medicinal plantTripterygiumwilfordiiHook f. (TWHF) whose extracts have been shown to have anti-inflammatory, immunosuppressive, as well as anti-cancer activity. It has been reported that rheumatoid arthritis, systemic lupus erythematosus, nephritis and psoriasis were successfully treated[1-3]. Recently, numerous breakthroughs have been made in our understanding of the anti-tumor activity of TP. Preclinical studies indicated that TP inhibits cell proliferation, induces cell apoptosis, inhibits tumor metastasis and enhances the effect of other therapeutic methods in various cancer cell lines. Several groups have demonstrated the potential value of TP to overcome chemoresistance in different types of cancer, including myeloid leukemia, pancreatic and ovarian cancer[2, 4-6].
Although TP possesses a variety of pharmacological effects bothinvitroandinvivo, it has been restricted in clinical application due to its multi-organ toxicity. The CFDA issued a warning in 2012 about this medicine, urging caution[7]. Clinically, the hepatotoxicity is one of the most significant adverse effects. Animal toxicology studies have shown that acute liver failure is one of the main causes of death associated with triptolide. The aggravated hepatotoxicity was evidenced with the lifted levels of alanine aminotransferase (ALT) and aspartate transaminase (AST) and remarkable histopathological changes, such as fat-like lesions and the swelling of the renal proximal tubule in the liver[8-10]. Our previous studies demonstrated that oxidative stress plays an important role in TP-induced liver injury in BALB/C mice[11]. Although many studies on the hepatotoxicity of TP have been reported, the underlying mechanism of TP-induced liver injury has not been fully elucidated.
The Notch pathway is an evolutionarily conserved fundamental pathway that regulates cell fate decisions in animals, such as cell differentiation, survival/apoptosis, and cell cycle[12]. It comprises four Notch receptors (Notch1-4) and two types of Notch ligands (Jagged1, 2 and Delta 1, 3, and 4) as well as intracellular proteins that transmit the Notch signal into the nucleus[13]. The binding of the Notch ligand to its re
ceptor triggers the γ-secretase-mediated proteolytic cleavage of the Notch intracellular domain (NICD). The NICD translocates into the nucleus, where it converts protein Jκ (RBPJκ) from a transcriptional repressor into an activator, which resulted in the transcription of Notch target genes such as hairy and enhancer of split 1 (Hes1) and the hairy-related transcription (HRT) factor family[14-15].
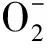
It is clear that the Nrf2 pathway is a dominant factor in the protection of cells against oxidative damage[17-18]. Nrf2 is evolutionarily conserved, as is Notch1, which indicated that cross-talk may occur between the two pathways. Several studies have shown that signaling in the direction of Notch1 to Nrf2 could be observedinvivoandinvitro.Insilicoanalyses and reporter gene assays revealed that RBPJκ recognition sequences were highly conserved in the Nrf2 promoter regions of mammals[19-20]. However, the precise mechanism underlying the combined action of Notch1-Nrf2 in the protection against hepatocyte damage is unknown.
This study was designed to determine the role of Notch1 signaling in TP-induced hepatotoxicity using Chang liver cells as aninvitromodel. To characterize the role of Notch1 in TP-induced hepatotoxicity, we suppressed or increased Notch1 expression and studied the downstream Nrf2 signaling because of these changes. Our findings suggested that the activation of Notch1 may offer a novel therapeutic strategy to protect against TP-induced hepatotoxicity.
1 Materials and methods
1.1 Reagents and antibodies
TP (>99% purity) was purchased from DND Pharm-Technology Co. (Shanghai, China). N-[N-(3,5-difluorophenacetyl)-L-alanyl]-S-phenylglycinet-butyl ester (DAPT, >98% purity) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Recombinant Human Jagged 1 Protein was purchased from R&D Company (Minneapolis, MN, USA). Anti-activated Notch1 was purchased from Abcam Biotechnology (Cambridge, MA, USA). Anti-Notch1 antibody and anti-HO-1 antibodies were purchased from Sangon Biotechnology (Shanghai, China). Anti-Bcl-2 antibody, anti-Caspase-3 antibody, and anti-Histone H3 antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Anti-Nrf2 antibody was purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). All other chemicals were of analytical grade from and available from commercial suppliers.
1.2 Cell culture and drug dissolution
Chang liver cells were obtained from American Type Culture Collection (ATCC, USA). The cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM, HyClone, Logan, UT, US) supplemented with 50 U·mL-1penicillin, 50 μg·mL-1streptomycin, and 10% fetal bovine serum (HyClone, Logan, UT, USA). The cells were grown in a humidified incubator in 5% CO2at 37 ℃. TP and DAPT stock solutions were prepared in dimethyl sulfoxide (DMSO) and were stored at -20 ℃. Jagged1 was dissolved in PBS and stored at -20 ℃.
1.3 Determination of intracellular GSH
To measure the generation of intracellular reduced GSH, Chang liver cells (8×104cells·well-1) were seeded in 6-well plates and then were exposed to TP (160 nmol/L) for 24 h. The GSH content was determined using a GSH Detection Kit (Beyotime, Jiangsu, China) in accordance with the manufacturer’s instructions.
1.4 Detection of apoptosis
Chang liver cells (8×104cells·well-1) were seeded in 6-well plates and were treated with TP (160
nmol/L) for 24 h. The Annexin V-FITC/PI Detection Kit (BD Biosciences, San Diego, CA) was used to determine the level of cellular apoptosis. Chang liver cells were exposed to TP for 24 h, harvested, washed twice with cold PBS, and resuspended in 1× binding buffer at a concentration of 1×106cells·mL-1. Subsequently, in accordance with the manufacturer’s instructions, the cells were stained with annexin V-FITC and PI for 20 min at 37 ℃, and analyzed immediately using a flow cytometer (Becton-Dickinson, San Jose, CA, USA).
1.5 Determination of ROS production
Chang liver cells (8×104cells·well-1) were seeded in 6-well plates and were treated with TP (160 nmol/L) for 24 h. After treatment, the cells were harvested and stained with 10 μmol/L 2′,7′-dichlorodihydrofluorescein diacetate (Sigma-Aldrich, St. Louis, MO, USA) at 37 ℃ for 20 min in the dark. The fluorescence was measured by using a flow cytometer (Becton-Dickinson, San Jose, CA, USA) measuring the FITC fluorescence; ROS was quantified using the mean fluorescence intensity.
1.6 Quantitative real-time PCR analysis
Total RNA was prepared using the RNAiso Plus (Takara, Japan), and the cDNA was prepared from the total RNA using the Prime Script RT reagent Kit (Takara, Japan) in accordance with the manufacturer’s instructions. The sequences of the primers are shown in Table 1. The PCR amplification was conducted on a LightCycler 2.0 system (Roche Diagnostics, Basel, Switzerland).
1.7 Gene knockdown using siRNA
Negative control siRNA, Notch1 siRNA (5′-UGGCGGGAAGUGUGAAGCG-3′ and 3′-ACCGCCCUUCACACUUCGC -5′) and Nrf2 siRNA (5′-GCAACAGGACAUUGAGCAA -3′and 3′-CGUUGUCCUGUAACUCGUU -5′) were obtained from RiboBio (Guangzhou, Guangdong, China). Chang liver cells were transiently transfected using Lipofectamine 3000 (Invitrogen, USA) in accordance with the manufacturer’s instructions. At 48 h after transfection, the cells processed and analyzed by western blot analysis.

Table 1 Primer sequences for PCR amplification
1.8 Western blot analysis
The Chang liver cell line was treated with different drugs. Then, the cells were harvested and washed with cold phosphate-buffered saline (PBS). The proteins were extracted with RIPA Cell Lysis Buffer (Beyotime, Jiangsu, China) and stored on ice for at least 20 min. The lysates were centrifuged at 12 000 ×gat 4 ℃ for 20 min, and the supernatant was transferred to a fresh tube. After the protein concentration was measured using the bicinchoninic acid (BCA) method, equal amounts of protein were loaded into each lane of a polyacrylamide gel, separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and transferred to polyvinylidene fluoride (PVDF) membranes. The membranes were blocked with 5% non-fat dry milk powder in 0.05% tris-buffered saline and Tween 20 (TBST) for 1 h at 25 ℃. After overnight incubation at 4 ℃ with antibodies, the membranes were washed three times and incubated for 1 h at 25 ℃ with secondary antibodies[21]. The membranes were developed using an electrochemiluminescence (ECL) kit (Thermo Scientific/Pierce, Rockford, IL, USA) in accordance with the manufacturer’s instructions or a chemiluminescence detection system (Bio-Rad Laboratories, Hercules, CA, USA). The density of the immunoreactive bands was analyzed by using ImageJ 1.41 (National Institutes of Health, Bethesda, MD, USA)[22].
1.9 Statistical analysis
The software SPSS v16.0 (SPSS Inc. Chicago, IL, USA) was used for statistical analysis. The data were expressed as the mean values ± S.D. The values were compared using Student’st-test and a one-way analysis of variance (ANOVA), followed by Tukey’s test. The levels of statistical significance were set atP<0.05, <0.01, orP<0.001.
2 Results
2.1 TP induces apoptosis and oxidative stress in Chang liver cells
It has been reported that TP induced cytotoxicity in both H9c2 cells and HepG2 cells in our previous work. TP reduced cell viability in a dose-and time-dependent manner. The IC50determined after 24 h exposure to TP was 157.87 nmol/L in HepG2 cells. In addition, 24 h incubation with various concentrations of TP markedly induced the release of LDH[18, 23, 24].
In our study, TP-induced hepatotoxicity was assessedinvitrousing Chang liver cells treated with various concentrations of TP for 24 h. As shown in Fig.1A, intracellular ROS increased by 200%, 260%, and 350% after addition of 80, 160, and 320 nmol/L TP, respectively. The cytotoxicity of TP was also assessed via the annexin V-FITC/PI double staining assay. Incubation with various concentrations of TP (80~320 nmol/L) for 24 h markedly increased the percentage of apoptotic cells (Fig.1B). These results indicated that oxidative stress and apoptosis were involved in the response of Chang liver cells to TP treatment.
2.2 TP suppresses activation of Notch1 pathway
Notch controls important aspects of liver homeostasis, metabolism, and vascular physiology. In a previous study, the conditional gene knockdown of Notch1 and overexpression of NICD confirmed that Notch1 and NICD were dominantly expressed in hepatocytes[20]. To explore the molecular mechanism underlying the TP-induced effects on Notch1 signaling, mRNA and protein expression of Notch1 signaling-related molecules were measured by qPCR and western blotting, respectively; additionally, NICD and Hes1 were analyzed as markers of Notch1 activation and downstream effector of Notch signaling, respectively.
Western blotting showed that NICD protein levels began to decrease 3 h after TP treatment. After 24 h of TP treatment, the values were 60% lower than those of the control group. Moreover, Notch1 protein levels began to decline at 6 h and reached the lowest value after 24 h of TP treatment (Fig.2A). In addition, after treatment with TP at various concentrations (0, 40, 80, 160, 320, and 640 nmol/L) for 24 h, NICD protein expression was lowest in the cells treated with 160 nmol/L. A gradual decline in the Notch1 protein level was also seen after treatment with various concentrations of TP for 24 h (Fig.2B). As shown in Fig.2C and D, Notch1 and Hes1 mRNA transcription in TP-treated cells was markedly decreased in a time-and dose-dependent manner, which suggested that Notch1 signaling was inhibited by TP. Collectively, these data suggested that TP significantly inhibited the Notch1 pathway in Chang liver cells.
2.3 Inhibition of Notch1 aggravates TP-induced cytotoxicity
DAPT, a specific inhibitor of γ-secretase, was shown to block proteolytic processing and reduce the release of NICD[25]. As shown in Fig.3A, after treatment with various concentrations of DAPT (0, 5, 10, 20 and 50 μmol/L) for 48 h, the NICD protein expression was lowest in the cells treated with 50 μmol/L. In our previous study, 160 nmol/L TP remarkably reduced the viability of HepG2 cells and H9c2 cells[18, 24]. Furthermore, 160 nmol/L TP significantly induced ROS production and apoptosis[7,18,26]. In the
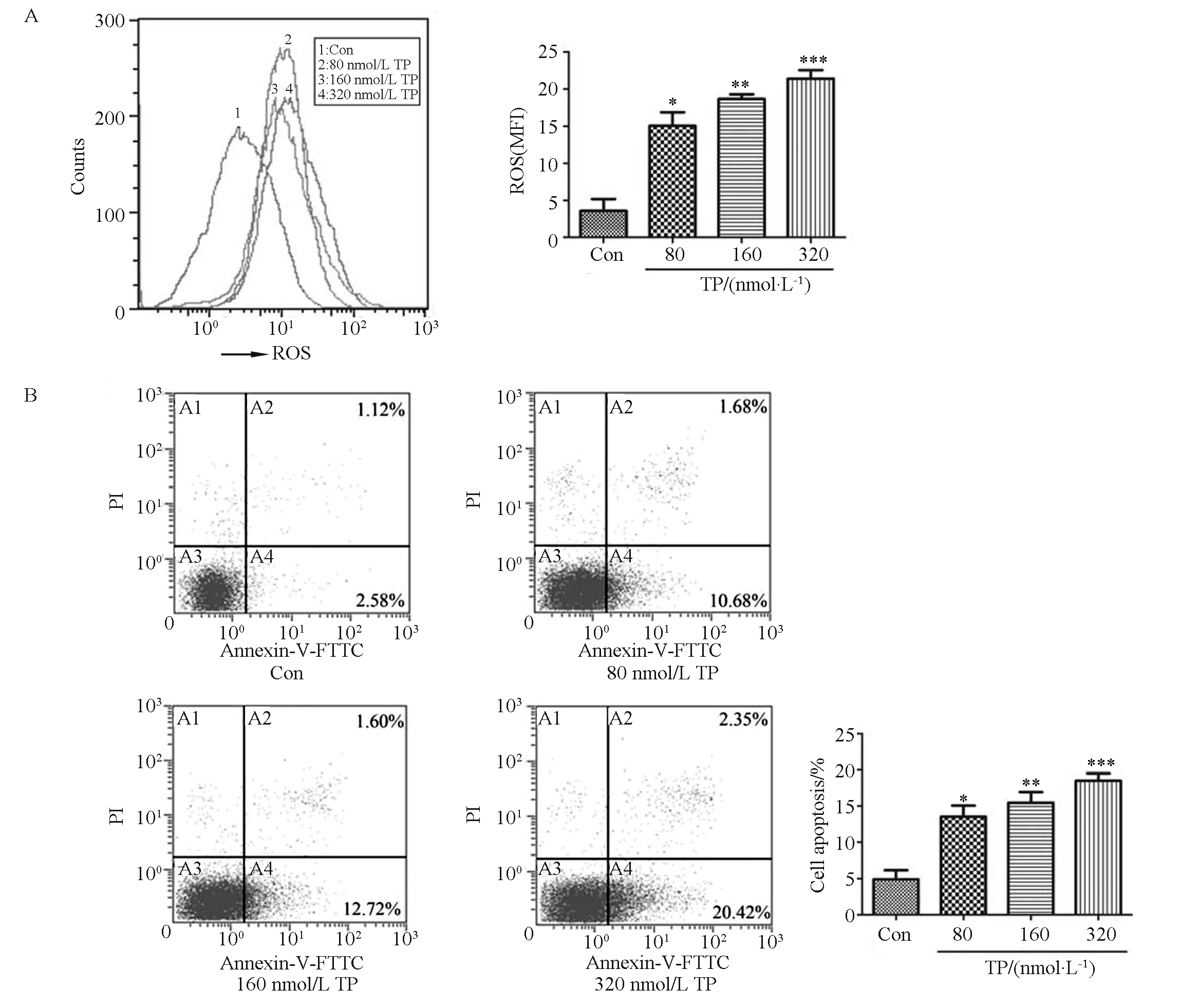
Fig.1 TP induces oxidative stress and apoptosis in Chang liver cells.Chang liver cells were treated with TP at various concentrations for 24 h. (A) ROS quantification was performed by using flow cytometry and the data are presented as the mean fluorescent intensity (MFI) after DCFH-DA staining; (B) The proportions of apoptotic cells were determined by annexin V-FITC/PI double staining. Data are presented as the mean ± SD from three independent experiments; *P < 0.05 vs control, ** P < 0.01 vs control, ***P < 0.001 vs control.
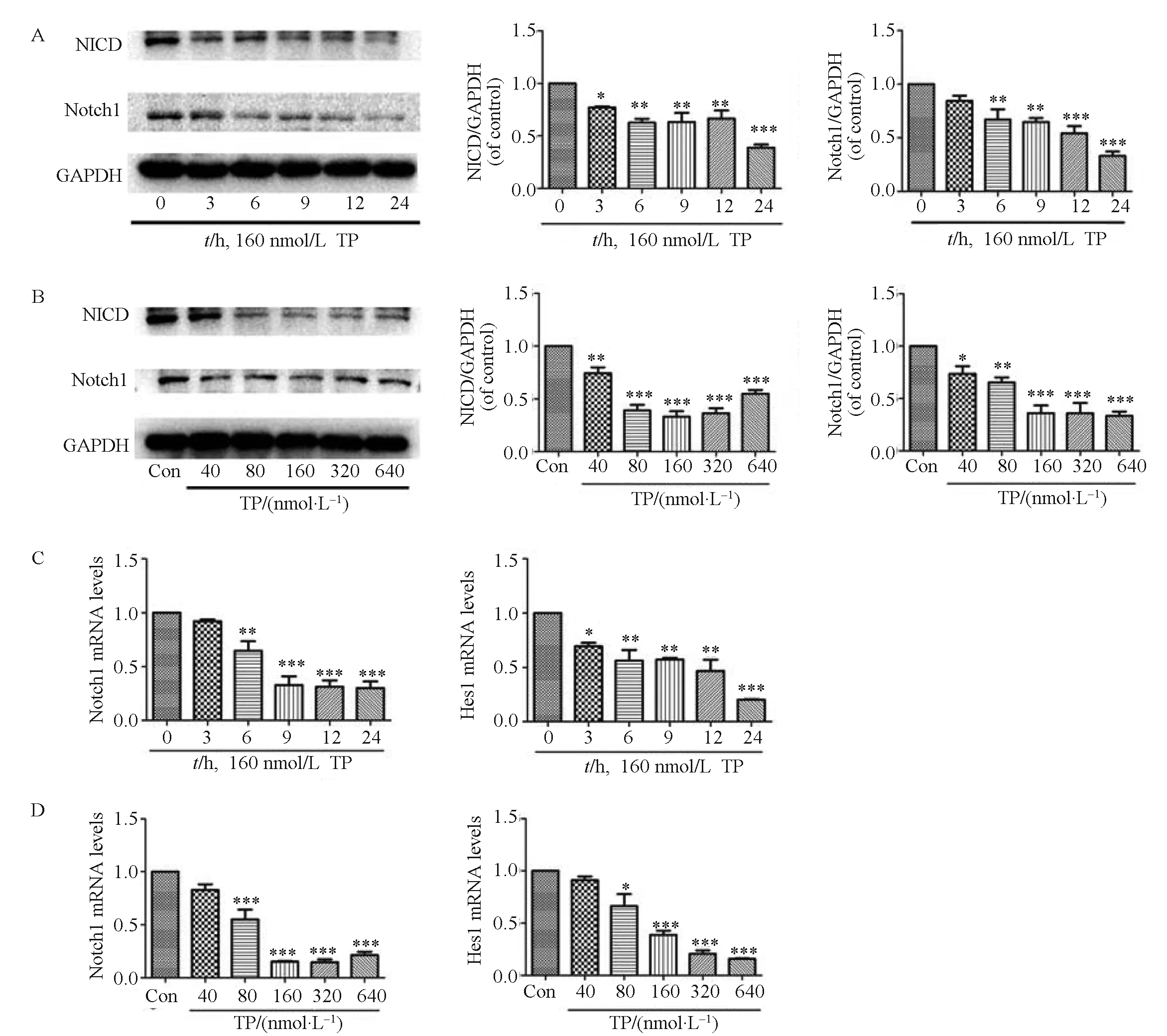
Fig.2 TP suppresses the activation of the Notch1 pathway. The effects of TP on the total levels of Notch1 and NICD were determined. Chang liver cells were treated with a fixed dose of TP (160 nmol/L) for different times (0, 3, 6, 9, 12 and 24 h) (A) or with various concentrations (0, 40, 80, 160, 320 and 640 nmol/L) of TP for 24 h (B). The time (C) and concentration (D) courses of mRNA expression of Notch1 and Hes1 in Chang liver cells were also analyzed. Data are presented as the mean ± SD from three independent experiments. *P < 0.05 vs control, ** P < 0.01 vs control, ***P < 0.001 vs control
present study, the connection between the Notch1 pathway and TP-induced cytotoxicity was investigated by pretreating Chang liver cells with DAPT (50 μmol/L) only for 24 h and then co-treatment with TP (160 nmol/L) and DAPT for 24 h. NICD expression was effectively decreased by DAPT treatment, as shown in Fig.3B. More importantly, when Chang liver cells were treated with both TP and DAPT, the expression of NICD decreased to a minimum, which indicated that DAPT enhanced the downregulation of Notch1 signaling after TP treatment.
Oxidative stress is known to contribute to TP-induced hepatotoxicity[9,24,27,28]. Therefore, we assessed oxidative injury by the measurement of ROS production and the level of cellular GSH, which is a key free radical scavenger in the protection against the oxidative processes in cells (Fig.3C and D). As illustrated in Fig.3C, after treatment with both TP and DAPT, the intracellular ROS level in Chang liver cells was approximately 1.5-fold higher than that in TP-treated cells. Additionally, DAPT treatment significantly enhanced the TP-induced depletion of GSH (Fig.3D).

Fig.3 Inhibition of Notch1 aggravates TP-induced cytotoxicity. Chang liver cells were treated with DAPT at various concentrations (0, 5, 10, 20, 50 μmol/L) for 48 h. (A) Western blotting analysis of NICD expression. Chang liver cells were pretreated with DAPT (50 μmol/L) for 24 h and then co-treated with TP (160 nmol/L) and DAPT for 24 h; (B) Western blotting analysis of NICD expression; (C) ROS quantification was performed by using flow cytometry and the data are presented as the mean fluorescent intensity (MFI) after DCFH-DA staining; (D) The level of GSH was determined by a GSH assay kit; (E) The percentages of apoptotic cells were determined by annexin V-FITC/PI double staining; (F) The apoptosis-related proteins were also determined by western blotting analysis. Chang liver cells were transiently transfected with negative control siRNA (NC), or siRNA targeting Notch1 for 48 h; (G) Western blot analysis of Notch1 expression. Chang liver cells were treated with TP (160 nmol/L) for 24 h after transfection with Notch1 siRNA; (H) Western blotting analysis of NICD expression; (I) The total levels of Bcl-2 and cleaved-caspase-3 were also assessed by western blotting; Data are presented as the mean ± SD from three independent experiments. *P < 0.05 vs control, **P < 0.01 vs control, ***P < 0.001 vs control; #P < 0.05 vs control cells treated with the same concentration of TP, ##P < 0.01 vs control cells treated with the same concentration of TP, ###P < 0.001 vs control cells treated with the same concentration of TP
The evaluation of DAPT-treated cells showed that the downregulation of NICD expression increased their susceptibility to TP-induced apoptosis, as indicated by a higher percentage of apoptotic cells, higher expression of cleaved-caspase-3, and lower expression of Bcl-2 (Fig.3E and F). These results indicated that the Notch1 signal blockade exacerbated TP-induced hepatocyte damage.
The role of Notch1 in TP-induced cytotoxicity was also evaluated in Chang liver cells after transfection with Notch1 siRNA. As shown in Fig. 3G, Chang liver cells were transiently transfected with siRNA targeting Notch1, negative control siRNA (NC), and vehicle control. We selected the third siRNA sequence as the following experimental tool. Notch1 siRNA successfully suppressed NICD protein expression levels by 65% compared with the vehicle under normal culture conditions (Fig.3H). Western blotting revealed that TP-treated cells transfected with Notch1 siRNA demonstrated a two-fold reduction in Bcl-2 expression compared with the TP group. In contrast, the Notch1 siRNA-transfected cells had a higher content of cleaved-caspase-3 after TP treatment (Fig.3I). In summary, these findings provided important evidence that interference with Notch1 expression exacerbated TP-induced apoptosis in Chang liver cells.
2.4 Activation of Notch1 by Jagged1 attenuates TP-induced cytotoxicity
To confirm the involvement of the Notch1 pathway in the protection against TP-induced hepatotoxicity, Chang liver cells were pretreated with Jagged1 (200 ng ·mL-1) only for 24 h and co-treated with TP (160 nmol/L) and Jagged1 for 24 h. As expected, incubation with Jagged1 increased the NICD protein level in Chang liver cells by approximately 1.7-fold compared with the control group (Fig.4A).
We hypothesized that Notch1 suppressed oxidative injury after TP treatment. We quantified intracellular ROS using the fluorescent probe DCFH-DA and evaluated cellular GSH levels in Chang liver cells (Fig.4B and C). The levels of intracellular ROS suggested that Jagged1 treatment effectively reduced TP-dependent ROS production, which was consistent with the enhanced levels of GSH observed.
The results of the annexin V-FITC/PI double-staining assays also indicated that treatment with both Jagged1 and TP reduced the apoptosis rate by approximately 43% compared with the TP group (Fig.4D). Western blot analysis showed that the pre-treatment with Jagged1 markedly attenuated TP-induced cellular apoptotic signaling by increasing the expression of Bcl-2 and decreasing the content of cleaved-caspase-3(Fig.4E).
2.5 Downregulation of Notch1 signaling inhibits Nrf2 activation and upregulation of Notch1 signaling enhances Nrf2 activation during TP-induced hepatotoxicity
Nrf2 is a direct transcriptional target of Notch1[20]; however, the precise mechanism underlying the combined action of Notch1 and Nrf2 in the protection of TP-induced cytotoxicity is unknown. To explore the impact of Notch1 signaling on Nrf2 activity, we detected the protein abundance of total cellular Nrf2, nuclear Nrf2, and HO-1 in Chang liver cells by western blotting.
The treatment with DAPT (50 μmol/L) decreased the expression of total cellular-Nrf2, nuclear Nrf2, and HO-1 by 33%, 47%, and 30% compared with the TP group, respectively (Fig.5A). Similarly, Notch1 siRNA effectively enhanced the TP-induced inhibition the expression of Nrf2-related molecules (Fig.5B). In contrast, treatment with Jagged1 increased the protein expression of total cellular Nrf2, nuclear Nrf2, and HO-1 by 1.9, 1.7, and 1.4-fold compared with the TP group, respectively, which indicated that the activation of Notch1 effectively counteracted the TP-induced inhibition of Nrf2 signal (Fig.5C). Collectively, these results provided additional evidence for cross-talk between the Notch1 and Nrf2 signaling pathways in Chang liver cells during TP-induced cytotoxicity.
2.6 Knockdown of Nrf2 attenuates Notch1 protection against TP-induced cytotoxicity
To investigate whether the Nrf2 pathway was essential in the hepatoprotective ability of Notch1, Chang liver cells were treated with TP (160 nmol/L) for 24 h after the transfection of Nrf2 siRNA in the presence of Jagged1. As shown in Fig. 6A, Chang liver cells were transiently transfected with siRNA targeting Nrf2, negative control siRNA (NC), and vehicle control. We
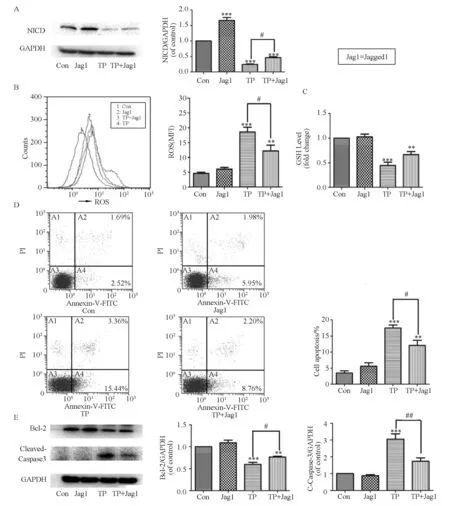
Fig.4 Activation of Notch1 by Jagged1 attenuates TP-induced cytotoxicity. Chang liver cells were pretreated with Jagged1 (200 ng·mL-1) for 24 h and then co-treated with TP (160 nmol/L) and Jagged1 for 24 h. (A) Western blotting analysis of NICD expression; (B) ROS quantification was performed using flow cytometry and the data were presented as the mean fluorescent intensity (MFI) after DCFH-DA staining; (C) The level of GSH was determined by a GSH assay kit; (D) The percentages of apoptotic cells were determined by annexin V-FITC/PI double staining; (E) The apoptotic-related proteins were also determined by Western blotting analysis; Data are presented as the mean ± SD from three independent experiments. **P < 0.01 vs control, ***P < 0.001 vs control; #P < 0.05 vs control cells treated with the same concentration of TP, ##P< 0.01 vs control cells treated with the same concentration of TP
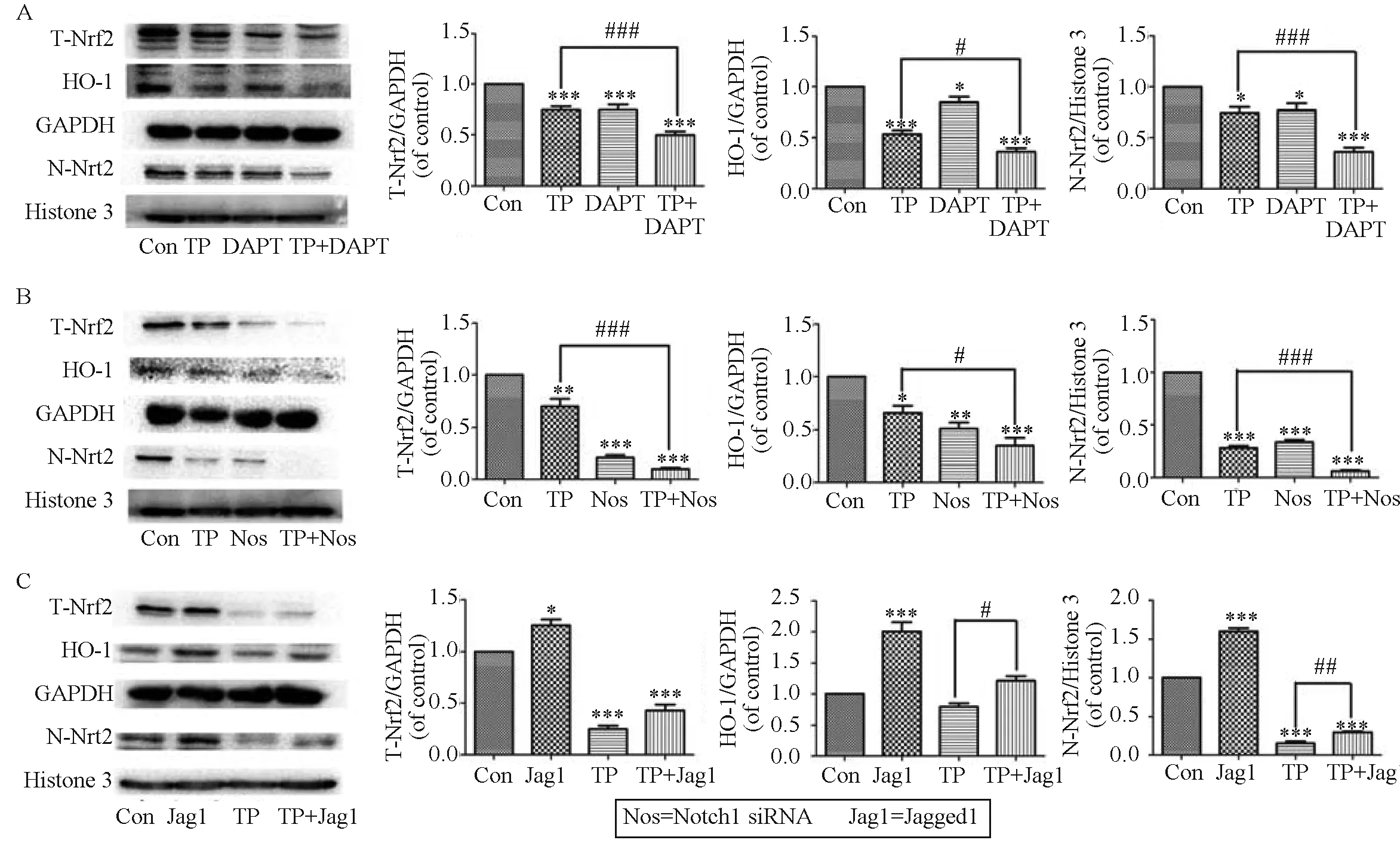
Fig.5 Downregulation of Notch1 signaling inhibits Nrf2 activation and upregulation of Notch1 signaling enhances Nrf2 activation during TP-induced hepatotoxicity. Chang liver cells were pretreated with DAPT (50 μmol/L) for 24 h and then co-treated with TP and DAPT for 24 h. (A) The levels of total cellular Nrf2, HO-1, and nuclear Nrf2 were analyzed. Chang liver cells were treated with TP (160 nmol/L) for 24 h after transfection with Notch1 siRNA; (B) The levels of total cellular Nrf2, HO-1, and nuclear Nrf2 were assessed by western blotting analysis. Chang liver cells were pretreated with Jagged1 (200 ng ·mL-1) for 24 h and then co-treated with TP and Jagged1 for 24 h; (C) The effects of Jagged1 on the expression levels of Nrf2 and its downstream proteins were determined; Data are presented as the mean ± SD from three independent experiments. *P < 0.05 vs control, **P < 0.01 vs control, ***P < 0.001 vs control; #P < 0.05 vs control cells treated with the same concentration of TP, ##P < 0.01 vs control cells treated with the same concentration of TP, ###P < 0.001 vs control cells treated with the same concentration of TP
chose the third siRNA sequence as the following experiment tool. As shown in Fig.6B, the antioxidative effect of Jagged1 was partially abrogated after Nrf2 siRNA transfection during TP-induced oxidative stress, which was supported by increased DCF fluorescence intensity. In agreement with this finding, Nrf2 deficiency mostly eliminated the ability of Jagged1 to protect against TP-induced apoptosis (Fig.6C). Collectively, these results illustrated that the Nrf2 pathway was crucial to maintain the actions of the Notch1 pathway in the promotion of hepatocyte survival after TP treatment. The key processes influenced by this protective mechanism may be the cycle of damage involving ROS overload and apoptosis.
3 Discussion
As the dominant active component of the Chinese herb TWHF, TP has been reported to be one of the most effective anti-inflammatory and immunomodulatory natural products ever discovered[22]. However, TP overdose can result in severe hepatotoxicity, and even death, in both animals and humans[23]. Therefore, the development of therapeutic intervention strategies to alleviate TP-induced toxicity would be of great clinic value.
Our previous studies indicated that the mechanisms underlying TP-induced toxicity mainly involved the regulation of oxidative stress[7, 18, 24, 26]. It has been demonstrated that TP treatment significantly increased the accumulation of ROS and subsequently triggered cellular apoptosis by depolarization of the mitochondrial membrane potential, decrease in the ratio of Bax/Bcl-2, release of cytochromec, and activation of caspase-3 in cardiomyocytes[18]. In this study, upregulated intracellular ROS production (Fig.1A) was observed in Chang liver cells after TP treatment. In addition, we found that TP significantly increased cell apoptosis (Fig.1B). These results indicated that oxidative stress and apoptosis were involved in the response of hepatocytes to TP treatment.
The Notch1 signaling pathway regulates various cellular events, such as cell growth, survival, and apoptosis[29]. Recent studies have demonstrated that the activation of Notch1 signaling may protect against apoptosis. For example, the administration of the Notch inhibitor may cause caspase-3-dependent apoptosis in adult islets and the activation of Notch1 protected against apoptosis[30]. Another study indicated that ischemia reperfusion-induced necrosis and apoptosis in cardiomyocytes were attenuated by activated Notch1 signaling[31]. Furthermore, a critical role of Notch1 signaling has been established in the protection against oxidative stress. A recent study found that Notch1 signaling mitigated oxidative stress during myocardial ischemia/reperfusion injury by rescuing cardiac thioredoxin system[16].
In order to investigate the mechanism underlying the TP-mediated inhibition of the Notch1 signaling pathway, we systematically evaluated the expression of Notch1, NICD, and Hes1 (Fig. 2A-D). We found
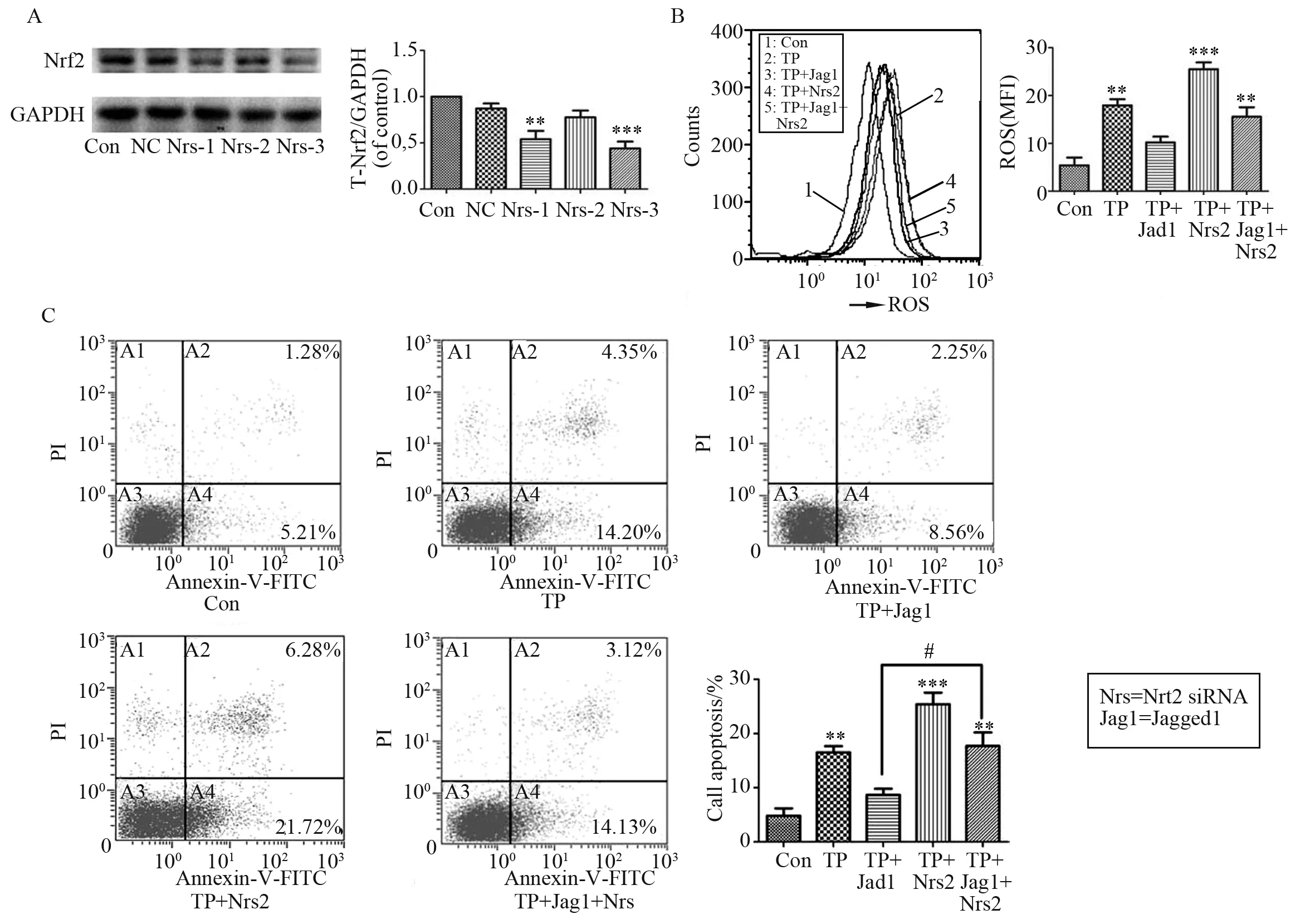
Fig.6 Knockdown of Nrf2 attenuates the protection of Notch1 against TP-induced cytotoxicity. Chang liver cells were transiently transfected with negative control siRNA (Con) or siRNA targeting Nrf2 for 48 h. (A) Western blot analysis of Nrf2 expression. Chang liver cells were treated with TP (160 nmol/L) for 24 h after Nrf2 siRNA transfection in the present of Jagged1; (B) ROS quantification was performed using flow cytometry and the data are presented as the mean fluorescent intensity (MFI) after DCFH-DA staining; (C) The percentages of apoptotic cells were determined by annexin V-FITC/PI double staining; Data are presented as the mean ± SD from three independent experiments. **P < 0.01 vs control, ***P < 0.001 vs control; #P < 0.05 vs control cells treated with the same concentration of TP and Jagged1
that TP not only decreased the mRNA and protein levels of Notch1, but also inhibited the accumulation of the active form of Notch1, NICD. The disturbance of Notch1/NICD signal transduction could be an important mechanism of the decreased mRNA level of Hes1 in Chang liver cells.
Furthermore, the data from our present study demonstrated that the downregulation of the Notch1 signaling resulted in increased intracellular ROS (Fig.3C) and depletion of GSH (Fig.3D), which suggested that the Notch1 signal blockade aggravated TP-induced oxidative stress in hepatocytes. The activation of Notch1 by Jagged1 resulted in the opposite effect (Fig.4B and C). Moreover, the pharmacological blockade of Notch1 promoted TP-induced cell apoptosis (Fig.3E and F), whereas the activation of Notch1 protected against TP-induced apoptosis (Fig.4D and E).
We elucidated the relationship between TP and Notch1 signaling in Chang liver cells. First, the expression of Notch1-related molecules was significantly reduced after TP treatment in Chang liver cells. In contrast, TP-induced oxidative stress and cellular apoptosis in Chang liver cells was markedly enhanced when Notch1 was blocked by DAPT, but was reduced when Notch1 was activated by Jagged1. Therefore, we concluded that Notch1 signaling exerted a protective effect on TP-induced oxidative damage and cellular apoptosis.
The nuclear transcription factor Nrf2 controls the expression and induction of defensive genes encoding detoxifying enzymes and antioxidant proteins; HO-1 is one of these defensive genes[32]. Our previous studies indicated that Nrf2 signaling played a key role in the protection against TP-induced hepatotoxicity, cardiotoxicity, and nephrotoxicity[18, 22, 24].
Notch1 and Nrf2 signaling pathways may exert a significant protective effect on TP-induced cytotoxicity by the mediation of levels of intracellular ROS and apoptosis-related molecules. Therefore, previous studies have raised an interesting question of whether the activation of the Nrf2 signal by Notch1 occurred during TP-induced cytotoxicity. In the present study, we found that the inhibition of Notch1 by DAPT or Notch1 siRNA significantly downregulated Nrf2 signaling (Fig.5A and B), whereas the Notch1 ligand induced the expression of Nrf2 and Nrf2 target genes (Fig.5C), which suggested cross-talk between Notch1 and Nrf2 in Chang liver cells.
To further elucidate a specific function for Nrf2, Chang liver cells were treated with TP (160 nmol/L) for 24 h after transfection with Nrf2 siRNA in the present of Jagged1. These results supported the notion that Nrf2 deficiency attenuated the ability of Jagged1 to protect against TP-induced oxidative stress (Fig.6B) and cellular apoptosis (Fig.6C). Therefore, we provided strong evidence that Nrf2 was essential for protection against the TP-induced cytotoxicity promoted by Notch1, which possibly occurred through the disruption of cellular apoptosis and ROS generation.
Several drugs and hormones were reported to exert protective effects in cardiac injury through the activation of Notch1 signaling. For example, berberine significantly improved cardiac function recovery and decreased myocardial apoptosis, infarct size, serum creatine kinase, and lactate dehydrogenase through the activation of the Notch1/Hes1 signaling pathway[33]. Relaxin significantly increased cell viability, decreased apoptosis, and reduced nitroxidative damage in cardiac muscle cells. These effects were attributable to the ability of relaxin to upregulate Notch1 signaling[34]. In addition, melatonin supplementation effectively ameliorated myocardial ischemia-reperfusion (MI/R) injury by the activation of Notch1/Hes1/Akt signaling and maintenance of the antioxidant balance in these circumstances[35]. However, it is unclear whether these materials confer a protective effect in liver injury. Therefore, we will research this topic in greater depth in the future.
There are several limitations of our study. The mechanisms that influence ROS production or apoptosis after the regulation of the cross-talk between Notch1 and Nrf2 are quite complex. Therefore, it is clear that Notch1-Nrf2 cross-talk produced a positive effect on TP-induced cytotoxicity. Nevertheless, several studies have indicated that Nrf2-Notch cross-talk influenced the expression of defense systems against various stressors, which enhanced the maintenance of cellular homeostasis[20, 36]. Therefore, we plan to examine the effects of Nrf2 on Notch1 signaling in TP-induced cytotoxicity in our future research. In addition, we consider there is a requirement forinvivostudies investigating the cross-talk between Notch1 and Nrf2 pathways during TP-induced toxicityinvivo.
4 Conclusion
Several lines of evidence from this study indicated that the Notch1 pathway was significantly inhibited in Chang liver cells following TP treatment. In addition, Notch1 signaling exerted an endogenous protective effect on TP-induced hepatotoxicity through an up-regulation of Bcl-2 expression and cellular antioxidant capacity, decrease in the level of cleaved-caspase-3, and suppression of the intracellular ROS generation. Although further study is needed to investigate the detailed mechanisms underlying the interaction between Notch1 and Nrf2 pathways, we proved that Notch1 signaling played a key role in the protection of TP-induced cytotoxicity and regulation of Nrf2 signaling transduction, which is the possible downstream signaling pathway involved in this process.
[1] ZIAEI S, HALABY R. Immunosuppressive, anti-inflammatory and anti-cancer properties of triptolide: A mini review [J]. Avicenna J Phytomed, 2016, 6 (2) : 149-164.
[2] ZHAO H, SHI P, DENG M, et al. Low dose triptolide reverses chemoresistance in adult acute lymphoblastic leukemia cells via reactive oxygen species generation and DNA damage response disruption [J]. Oncotarget, 2016, 7 (51) : 85515-85528.
[3] HAN R, ROSTAMI-YAZDI M, GERDES S, et al. Triptolide in the treatment of psoriasis and other immune-mediated inflammatory diseases [J]. British Journal of Clinical Pharmacology, 2012, 74 (3) : 424-436.
[4] MENG C, ZHU H, SONG H, et al. Targets and molecular mechanisms of triptolide in cancer therapy [J]. Chinese Journal of Cancer Research, 2014, 26 (5) : 622-626.
[5] WANG C, LIU B, XU X, et al. Toward targeted therapy in chemotherapy-resistant pancreatic cancer with a smart triptolide nanomedicine [J]. Oncotarget, 2016, 7 (7) : 8360-8372.
[6] ZHONG Y Y, CHEN H P, TAN B Z, et al. Triptolide avoids cisplatin resistance and induces apoptosis via the reactive oxygen species/nuclear factor-kappaB pathway in SKOV3PT platinum-resistant human ovarian cancer cells [J]. Oncology Letters, 2013, 6 (4) : 1084-1092.
[7] WANG W, YANG Y, XIONG Z, et al. Inhibition of glycogen synthase kinase 3beta ameliorates triptolide-induced acute cardiac injury by desensitizing mitochondrial permeability transition [J]. Toxicology and Applied Pharmacology, 2016, 313: 195-203.
[8] QU L, QU F, JIA Z, et al. Integrated targeted sphingolipidomics and transcriptomics reveal abnormal sphingolipid metabolism as a novel mechanism of the hepatotoxicity and nephrotoxicity of triptolide [J]. Journal of Ethnopharmacology, 2015, 170: 28-38.
[9] FU Q, HUANG X, SHU B, et al. Inhibition of mitochondrial respiratory chain is involved in triptolide-induced liver injury [J]. Fitoterapia, 2011, 82 (8) : 1241-1248.
[10] SHEN G, ZHUANG X, XIAO W, et al. Role of CYP3A in regulating hepatic clearance and hepatotoxicity of triptolide in rat liver microsomes and sandwich-cultured hepatocytes [J]. Food and Chemical Toxicology, 2014, 71: 90-96.
[11] LI J, SHEN F, GUAN C, et al. Activation of Nrf2 Protects against triptolide-Induced hepatotoxicity [J]. PLoS One, 2014, 9 (7) : e100685.
[12] BI P, KUANG S. Notch signaling as a novel regulator of metabolism [J]. Trends in Endocrinology and Metabolism, 2015, 26 (5) : 248-255.
[13] ANDROUTSELLIS-THEOTOKIS A, LEKER R R, SOLDNER F, et al. Notch signalling regulates stem cell numbers in vitro and in vivo [J]. Nature, 2006, 442 (7104) : 823-826.
[14] BRAY S J. Notch signalling: a simple pathway becomes complex [J]. Nature Reviews Molecular Cell Biology, 2006, 7 (9) : 678-689.
[15] LAI E C. Notch signaling: control of cell communication and cell fate [J]. Development, 2004, 131 (5) : 965-973.
[16] PEI H, YU Q, XUE Q, et al. Notch1 cardioprotection in myocardial ischemia/reperfusion involves reduction of oxidative/nitrative stress [J]. Basic Research in Cardiology, 2013, 108 (5) : 373.
[17] NGUYEN T, YANG C S, PICKETT C B. The pathways and molecular mechanisms regulating Nrf2 activation in response to chemical stress [J]. Free Radic Biol Med, 2004, 37 (4) : 433-441.
[18] ZHOU J, XI C, WANG W, et al. Triptolide-induced oxidative stress involved with Nrf2 contribute to cardiomyocyte apoptosis through mitochondrial dependent pathways [J]. Toxicology Letters, 2014, 230 (3) : 454-466.
[19] WAKABAYASHI N, SKOKO J J, CHARTOUMPEKIS D V, et al. Notch-Nrf2 axis: regulation of Nrf2 gene expression and cytoprotection by notch signaling [J]. Mol Cell Biol, 2014, 34 (4) : 653-663.
[20] WAKABAYASHI N, CHARTOUMPEKIS D V, KENSLER T W. Crosstalk between Nrf2 and Notch signaling [J]. Free Radic Biol Med, 2015, 88 (Pt B) : 158-167.
[21] CHEN X, TAN M, XIE Z, et al. Inhibiting ROS-STAT3-dependent autophagy enhanced capsaicin-induced apoptosis in human hepatocellular carcinoma cells [J]. Free Radic Res, 2016, 50 (7) : 744-755.
[22] LI J, JIN J, LI M, et al. Role of Nrf2 in protection against triptolide-induced toxicity in rat kidney cells [J]. Toxicology Letters, 2012, 213 (2) : 194-202.
[23] JIN J, SUN X, ZHAO Z, et al. Activation of the farnesoid X receptor attenuates triptolide-induced liver toxicity [J]. Phytomedicine, 2015, 22 (10) : 894-901.
[24] LI J, SHEN F, GUAN C, et al. Activation of Nrf2 protects against triptolide-induced hepatotoxicity [J]. PLoS One, 2014, 9 (7) : e100685.
[25] XIAO Y G, WANG W, GONG D, et al. gamma-Secretase inhibitor DAPT attenuates intimal hyperplasia of vein grafts by inhibition of Notch1 signaling [J]. Lab Invest, 2014, 94 (6) : 654-662.
[26] YANG Y, WANG W, XIONG Z, et al. Activation of SIRT3 attenuates triptolide-induced toxicity through closing mitochondrial permeability transition pore in cardiomyocytes [J]. Toxicology in vitro, 2016, 34: 128-137.
[27] CAO L J, LI H D, YAN M, et al. The Protective Effects of Isoliquiritigenin and Glycyrrhetinic Acid against Triptolide-Induced Oxidative Stress in HepG2 Cells Involve Nrf2 Activation [J]. Evidence-Based Complementary and Alternative Medicine, 2016, 2016: 8912184.
[28] WANG J, JIANG Z, JI J, et al. Gene expression profiling and pathway analysis of hepatotoxicity induced by triptolide in Wistar rats [J]. Food and Chemical Toxicology, 2013, 58: 495-505.
[29] MO J S, YOON J H, ANN E J, et al. Notch1 modulates oxidative stress induced cell death through suppression of apoptosis signal-regulating kinase 1 [J]. Proc Natl Acad Sci U S A, 2013, 110 (17) : 6865-6870.
[30] DROR V, NGUYEN V, WALIA P, et al. Notch signalling suppresses apoptosis in adult human and mouse pancreatic islet cells [J]. Diabetologia, 2007, 50 (12) : 2504-2515.
[31] ZHOU X L, WAN L, XU Q R, et al. Notch signaling activation contributes to cardioprotection provided by ischemic preconditioning and postconditioning [J]. J Transl Med, 2013, 11: 251.
[32] KASPAR J W, NITURE S K, JAISWAL A K. Nrf2:INrf2 (Keap1) signaling in oxidative stress [J]. Free Radic Biol Med, 2009, 47 (9) : 1304-1309.
[33] YU L, LI F, ZHAO G, et al. Protective effect of berberine against myocardial ischemia reperfusion injury: role of Notch1/Hes1-PTEN/Akt signaling [J]. Apoptosis, 2015, 20 (6) : 796-810.
[34] BOCCALINI G, SASSOLI C, FORMIGLI L, et al. Relaxin protects cardiac muscle cells from hypoxia/reoxygenation injury: involvement of the Notch-1 pathway [J]. FASEB J, 2015, 29 (1) : 239-249.
[35] YU L, FAN C, LI Z, et al. Melatonin rescues cardiac thioredoxin system during ischemia-reperfusion injury in acute hyperglycemic state by restoring Notch1/Hes1/Akt signaling in a membrane receptor-dependent manner [J]. Journal of Pineal Research, 2017, 62 (1) : 1-17.
[36] WAKABAYASHI N, SHIN S, SLOCUM S L, et al. Regulation of notch1 signaling by nrf2: implications for tissue regeneration [J]. Sci Signal, 2010, 3 (130) : ra52.
2017-02-22 基金项目:国家自然科学基金(81473416);广东省科技计划项目(2016B020237003)
熊哲文 (1991年生),女;研究方向:药物毒理学;E-mail: xiongzhw5@mail2.sysu.edu.cn
黄芝瑛(1965年生),男;研究方向:药物毒理学;E-mail: hzhiying@mail.sysu.edu.cn 沈飞海(1980年生),男;研究方向:药物毒理学;E-mail: shenfh3@mail.sysu.edu.cn
R965
A
0529-6579(2017)03-0105-14
Notch1信号通路在雷公藤甲素所致肝细胞毒性的作用及机制
熊哲文,王丽,孔嘉敏,汪文文,程一森,沈飞海,黄芝瑛
(中山大学药学院,广东 广州 510006)
雷公藤甲素(triptolide, TP)是从植物雷公藤中分离得到的活性最高的二萜内酯类化合物。它具有抗炎、免疫调节、抗肿瘤等多种药理活性,目前在国内外被广泛研究与应用。然而,雷公藤甲素的肝脏毒性,限制了它的临床应用。为探讨Notch1信号通路对雷公藤甲素所致肝细胞毒性的保护作用及其相关机制,减轻雷公藤甲素肝脏毒性提供理论依据。研究表明,雷公藤甲素可以导致张氏肝细胞(Chang liver cell)的氧化应激水平及细胞凋亡水平增加,同时明显抑制Notch1信号通路的激活。此外,通过外源性给予Notch1配体Jagged1激活Notch1信号通路,可以保护雷公藤甲素诱导的氧化应激损伤及细胞凋亡损伤,在这个过程中,Nrf2及其下游靶点HO-1的表达水平增加。而通过给予DAPT或siRNA干扰Notch1抑制Notch1信号通路,则加重雷公藤甲素的细胞毒性,同时抑制Nrf2信号通路的激活。进一步的研究表明,siRNA干扰Nrf2可以部分消除Jagged1对雷公藤甲素所致肝细胞毒性的保护作用。以上实验结果表明,激活Notch1信号通路可以保护雷公藤甲素诱导的肝细胞氧化应激损伤及细胞凋亡损伤,而Nrf2是发挥该保护作用的重要下游靶点。
雷公藤甲素;Notch1;Nrf2;氧化应激;细胞凋亡
10.13471/j.cnki.acta.snus.2017.03.016
猜你喜欢
现代农药(2022年4期)2022-11-19
中草药(2022年17期)2022-09-05
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中华实用诊断与治疗杂志(2022年1期)2022-08-31
昆明医科大学学报(2021年6期)2021-07-31
中华养生保健(2020年9期)2021-01-18
中国现代中药(2020年9期)2020-11-16
药学研究(2015年11期)2015-12-19
科技传播(2014年4期)2014-08-15
山东农药信息(2013年1期)2013-03-29
