
基于高通量测序的玄参根部转录组学研究及萜类化合物合成相关基因的挖掘
2017-07-31 08:28潘媛陈大霞宋旭红张雪李隆云
中国中药杂志 2017年13期
潘媛+陈大霞+宋旭红+张雪+李隆云


[摘要] 该研究应用新一代测序技术Illumina HiSeqTM4000在转录水平上对药用植物玄参根部进行测序,结合生物信息学方法开展基因功能注释和SSR位点搜索。通过测序,共获得65 602 036条原始序列。利用生物信息学软件拼接和组装序列,获得73 983条unigene,平均长度823 bp。序列同源性比较表明,56 389条unigene与其他物种具有不同程度的同源性。通过Swiss-Prot,GO,KEGG,COG比对注释,发现520条编码玄参次生代谢途径关键酶基因和191个相关转录因子。利用MISA软件在所有unigenes中共搜索到11 659个SSR位点,重复类型以二核苷酸为主。该研究所获得的参与次生代谢的关键基因可为研究玄参药用成分的生物合成和调控机制奠定基础,获得的大量SSR位点为后续研究玄参种质鉴定及遗传多样性研究提供参考。
[关键词] 玄参;转录组;高通量测序;萜类物质
[Abstract] To investigate the profile of gene function and search for SSR, a new technology of high-throughput Solexa / Illumina sequencing was used to generate the root transcriptome of Scrophularia ningpoensis, and 65 602 036 raw reads were obtained. Based on the bioinformatics analysis and Trinity, 73 983 unigenes were obtained with an average length of 823 bp. The comparison of sequence homology in database showed that 56 389 unigenes had different degrees of homology. A total of 520 metabolic pathways related genes and 191 related transcription factors were identified by the Swiss-Prot, GO, KEGG and COG.The 11 659 SSRs were found by MISA and the highest frequency was AG/CT. In this study, we obtained numerous SSRs to provide references for the study of functional gene cloning and genetic diversity of S. ningpoensis. The key genes involved in the secondary metabolism are the basis for the study of biosynthesis and regulatory mechanism of the secondary metabolites.
[Key words] Scrophularia ningpoensis;transcriptome;high throughput sequencing;terpenoids
玄參为玄参科植物玄参Scrophularia ningpoensis Hemsl.的干燥根。玄参为我国常用中药材,始载于《神农本草经》,列为中品,历代药典都有收载。味甘、苦、咸、微寒,具有清热凉血,滋阴降火,解毒散结等功效[1]。研究发现玄参含有环烯醚萜、苯丙素、多糖等多种化学成分,具有保护心脑血管系统、抗炎、增强免疫等药理活性[2]。长期以来,由于玄参分子生物学相关研究起步较晚,缺乏玄参生长发育相关的分子标记开发、遗传图谱构建以及次生代谢途径等基础性研究成果的支撑,玄参分子育种、药效成分合成研究进展缓慢。高通量测序技术的出现,为研究玄参生长发育及次生代谢的分子机制提供了重要的基因资源,并为开展玄参功能基因组学研究提供了全新的思路和方法[3-4]。
高通量转录组测序技术已广泛应用于生物体转录组基因表达分析,采用该技术能全面快速地获取研究对象在某一状态下基因转录信息,从中挖掘重要功能基因,揭示不同生物学性状的分子机制[5-7]。开展玄参转录组的研究,也可能发现一些与其药效活性成分生物合成相关的候选基因,为玄参药效资源的充分利用奠定基础。本研究拟在转录水平上,利用Illumina HiSeqTM4000测序技术构建玄参根系转录组数据库,获得玄参转录本信息,并进行功能注释及SSR位点分析,揭示玄参根系转录组的整体表达特征,为进一步揭示玄参有效成分的累积、道地性形成等生物学过程的分子生物学研究提供丰富的数据资源。
1 材料与方法
1.1 样品 药用植物玄参块根采自重庆市武隆县仙女山玄参GAP种植基地,采集时间为2015年8月初(块根膨大期),经重庆市中药研究院李隆云研究员鉴定为玄参科玄参属植物玄参S. ningpoensis。选择生长健壮无病害的玄参植株,纯水洗净整个块根,用灭菌后的吸水纸吸干表面水分,迅速将块根切成约5 mm厚的薄片,立即用液氮速冻,后放入-80 ℃冰箱保存备用。
1.2 RNA的提取与转录组测序 采用 Trizol Reagent (Invitrogen)法提取玄参根总RNA,使用Agilen2100生物分析仪和NanoDrop分光光度计对提取的总RNA进行质量检测。总RNA质检合格后,用带有Oligo (dT)的磁珠富集真核生物mRNA加入fragmentation buffer,将mRNA打断成短片段,以mRNA为模板,用6碱基随机引物(random hexamers)反转录合成第一条cDNA链,然后加入缓冲液、dNTPs、RNase H和DNA polymerase Ⅰ合成双链cDNA链,经过QiaQuick PCR试剂盒纯化并加EB缓冲液洗脱之后做末端修复、加poly (A)并连接测序接头,然后用琼脂糖凝胶电泳进行片段大小筛选,接着进行PCR扩增,构建好的文库用Illumina HiSeqTM4000进行测序。
1.3 数据的拼接与组装 经测序获得的原始序列(raw reads),去除里面含有带接头的、低质量的reads,评估测序数据质量,并对测序数据进行过滤,从而获得干净序列(clean reads)。本研究采用Trinity[8]对clean reads进行拼接。该软件通过序列之间的重叠(overlap)信息组装得到重叠群(contigs),然后局部组装得到转录本(transcripts),最后用 TGICL和 Phrap 软件对转录本进行同源聚类和拼接得到单基因簇(unigene)。
1.4 功能注释与分类 通过blastx将拼接所得unigene比对到Nr[10](Non-redundant protein database,非冗余蛋白数据库),Nt,Swiss-Prot(SwissProt protein database,蛋白质序列数据库),GO[11](Gene Ontology,基因本体论数据库)、KEGG(Kyoto Encyclopedia of Genes and Genomes,东京基因与基因组百科全书)和COG(Cluster of Orthologous Groups,蛋白质直系同源数据库) (e-value<10-5),从而获得该unigene的功能注释信息[9]和分类信息,对所有注释信息进行整理。
1.5 SSR位点筛选 将转录组数据用MISA 软件进行SSR分析。设置参数如下:总重复序列长度不低于20 bp;二核苷酸、三核苷酸、四核苷酸、五核苷酸和六核苷酸至少重复次数分别为10,7,5,4,4 [14]。
2 结果与分析
2.1 转录组测序与数据组装 采用Illumina HiSeqTM4000高通量测序技术对玄参根系转录组进行了测序,共得到6 560万条raw reads以及6 456万条clean reads。本研究clean reads Q20为97%(一般为>90%),GC量为44.82%,基本呈正态分布,质量合格。采用Trinity软件组装共产生109 260个转录本,平均长度为493 nt。一般把所有转录本中最长的一个视为unigene,共获得了73 983个unigene,长度201~15 502 nt,见表1。
2.2 序列功能注释与分类 使用BLAST程序将组装得到的unigene与NT,NR,KOG,GO,Swissprot,KEGG数据库进行比对,进行unigene的序列相似性分析,从而得到该unigene的蛋白质功能注释信息。其中,匹配到 NR数据库中的有56 389条,占全部unigene的76.21%,其后依次是Swissprot(56.44%),Nt(55.9%),GO(50.47),KO(31.05%),KOG(21.97%)。对这6种数据库进行拓扑分析,共有9 494条unigene在所有数据库中同时标注成功,占总unigene数的12.83%,并且在所有数据库中至少有1种数据库注释成功的unigene有58 948条,占总unigene数的79.67%。
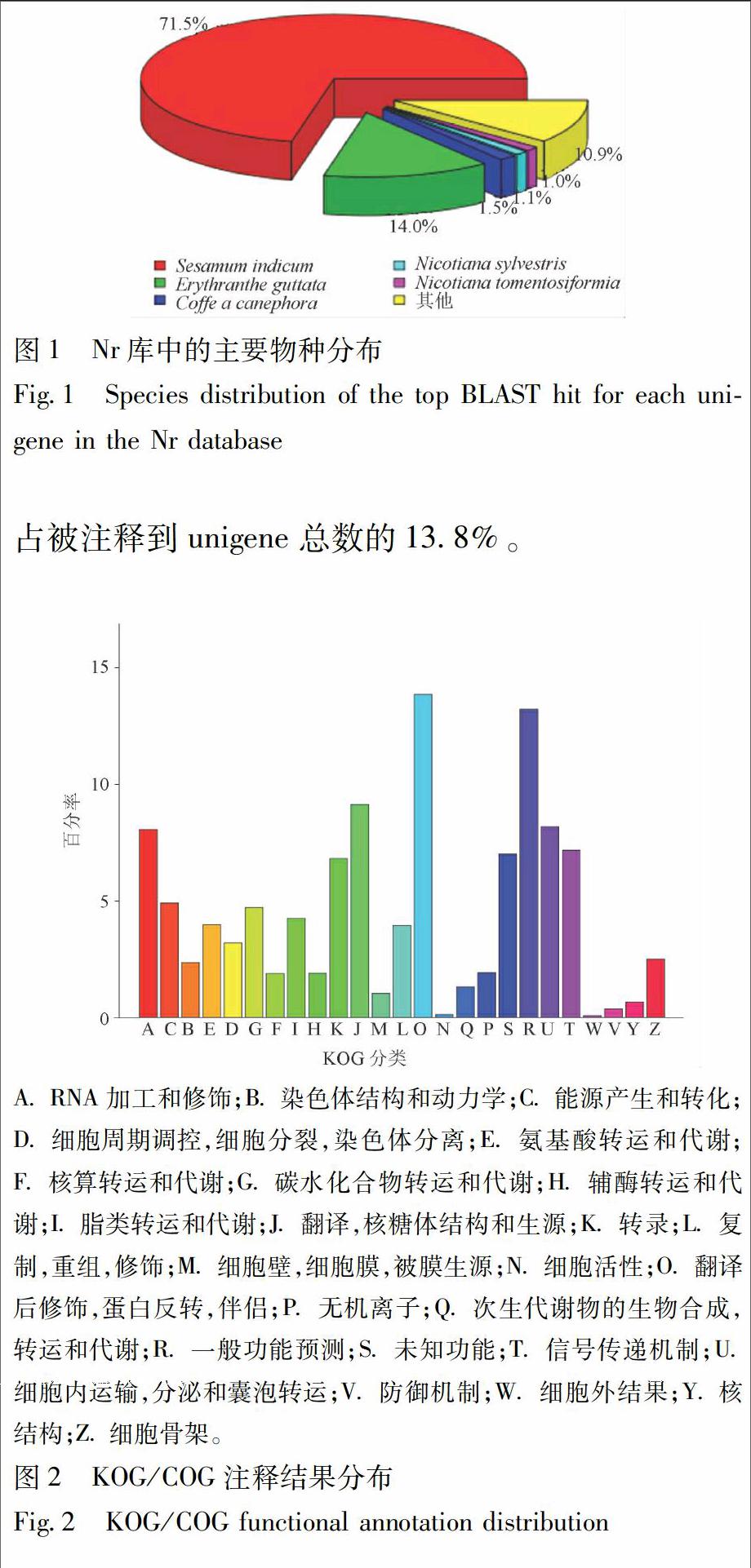
以 NR 数据库为例进行分析,56 389条unigenes在NR数据库中可找到相似序列。注释基因同源序列的物种分布情况见图1,在相似序列匹配度较高的近缘物种中,芝麻Sesamum indicum所占比例最高,为71.5%;其次是合瓣花Erythranthe guttata所占比例为14.0%,这些物种都为本研究中的序列注释提供了参考序列。
将玄参Unigene与KOG数据库进行对比,可预测 unigene功能并进行分类统计。结果表明,共有16 126条 unigene(14.66%)被注释到26种KOG分类中,见图2。从图中可以看出unigene涉及的KOG功能类别比较全面,涉及了大多数的生命活动。如RNA加工与修饰、能量的合成与运输、氨基酸转运与代谢、染色体结构和动力学等。其中,“翻译后修饰,蛋白质转运”是最大类别,包含2 233条unigene,结合GO数据库对玄参根系的unigene进行功能分类,可从宏观上认识玄参根系表达基因的功能分布特征。试验结果表明,有 37 346条unigene被注释到GO分类,其中参与生物学过程(biological process)分类中主要聚集于细胞过程(cellular process,21 126个),代谢过程(metabolic process,19 743个)和生物调节(biological regulation,7 192个);在细胞组分(cellular component) 主要聚集于细胞 (cell,9 369个)、细胞成分(cell part,9 364个)和细胞器(organelle,7 780个);在分子功能(molecular function)分类中主要聚集于结合蛋白(binding,21 810个)和催化活性(catalytic activity,16 641个),见图3。
2.3 序列代谢通路分析 根据KEGG数据库的注释信息能进一步得到unigene的代谢通路注释。本研究将unigene根据参与的KEGG代谢通路分为5个分支:细胞过程(A),环境信息处理(B),遗传信息处理(C),代谢(D)和有机系统(E),其中涉及较多的有遗传信息处理中的翻译(2 096个)、折叠、分类和降解(1 897条),涉及最少的是环境信息处理中的膜转运(91条),见图4。
结合KEGG数据库,对玄参根系的 unigene 可能参与或涉及的代谢途径进行了统计分析。结果表明,22 972条unigene参与到129个代谢通路中,与玄参次生代谢相关的unigene有782条。主要代谢产物有16種,这些代谢产物分别为花青素(anthocyanin)、咖啡因(caffeine)、黄酮和黄酮醇(flavone and flavonol)、类黄酮(flavonoid)、芥子油苷(glucosinolate)、异黄酮(isoflavonoid)、异喹啉类生物碱(isoquinoline alkaloid)、苯丙素(phenylpropanoid)、类固醇(steroid)、生物素(biotin)、油菜素内酯(brassinosteroid)、类胡萝卜素(carotenoid)、萜类化合物(terpenoid)、柠檬烯和蒎烯(limonene and pinene)和玉米素(zeatin)。玄参药用成分主要有环烯醚萜类、苯丙素、多糖、部分黄酮类等,其中注释到萜类、苯丙素类、黄酮类物质生物合成与代谢途径的unigene分别有56,249,52条,见图5。
2.4 玄参次生代谢途径相关基因的挖掘 环烯醚萜类、苯丙素类是玄参的主要药用成分,它们的生物合成和代谢涉及到细胞色素P450、DXR-1-脱氧-D-木酮糖-5-磷酸还原异构酶(1-deoxy-D-xylulose 5-phosphate reductoisomerase)、FPPS-法呢基焦磷酸合成酶(farnesyl pyrophosphate synthase )、HMGS-3-羟基-3-甲基戊二酰辅酶A(3-hydroxy-3-methyglutaryl-CoA)及HMGR-HMG-CoA还原酶等酶的作用[15],以上提到的酶都存在于玄参根中,其中编码细胞色素P450家族相关酶的unigene共搜索到504条,编码1-脱氧-D-木酮糖-5-磷酸还原异构酶的unigene共搜索到9条,法呢基焦磷酸合成酶共搜索到1条,3-羟基-3-甲基戊二酰辅酶A 和HMG-CoA还原酶各搜索到3条,见表2。
转录因子也称反式作用因子,是能够与真核基因启动子区域中顺式作用原件发生特异性相互作用的DNA结合蛋白,通过他们之间以及与其他相关蛋白之间的相互作用,激活或抑制转录。本研究使用iTAK软件对玄参转录组序列信息进行转录因子预测,发现有3 919条unigene分属于72个转录因子家族。目前发现的植物萜类转录因子主要包括AP2/ERF類、WRKY类、锌指类、bZIP类、bHLH类等[15]。在玄参转录组信息中与萜类合成相关的AP2/ERF类转录因子的表达丰度最高,涉及到的unigene有191条,见图6。AP2/ERF类转录因子是植物特有的一类转录因子,AP2/ERF家族成员在结构上含有一个或多个AP2/ERF结构域。每个AP2/ERF结合域有2个保守序列块—YRG原件和RAYD原件[16-18]。该转录因子已从拟南芥、烟草、水稻、玉米等多种植物中分离获得,他们在植物的生长、发育、各种生物和非生物胁迫以及多种生理生化反应中发挥重要作用。此外,WRKY类转录因子的表达丰度也较高,它是近年来新发现的植物特有的锌指型转录调控因子,能够调控植物信号转导和生理生化过程,调控植物次生代谢途径中编码关键酶基因的活性,并在植物抗病及免疫方面具有重要作用[19-20]。这些转录因子的发现将有助于玄参次生代谢成分生物合成途径的进一步研究。
2.5 SSR位点分析 SSR,简单重复序列标记(simple sequence repeats),又称为短串联重复序列或微卫星标记,是一类由几个核苷酸(1~6个)为重复单位组成的长达几十个核苷酸的重复序列,长度较短,且广泛均匀分布于真核生物基因组中。由于重复单位的核苷酸不同以及重复次数不完全相同,造成了SSR长度的高度变异性,其中最常见的双核苷酸重复类型,如(CA)n。一般采用SSR分子标记法对物种种质资源进行遗传多样性分析。本实验利用MISA软件在玄参根系的73 983条unigenes中共搜索到11 659个SSR位点,其中10 022条序列都存在SSR位点。SSR 的类型丰富,单核苷酸至六核苷酸重复类型均存在,所占比例变化较大,见表3。其中,二核苷酸重复所占比例最高,达到了40.13%;比例最低的是五核酸重复,仅为 0.20%;单核苷酸重复和三核苷酸重复所占比例大致相当,分别为30.46%,27.87%。在检测结果中,共出现61种基序类型,出现频率最高的6类基序为:AG/CT(2475),AT/AT(1316),AC/GT(885),ATC/ATG(692),AAG/CTT(590)和ACC/GGT(572)。上述 SSR 特征分析,有助于开展玄参及其同属物种的基因组差异分析、分子标记开发和遗传连锁图谱构建的研究。
3 讨论
目前,高通量转录组测序技术已经广泛应用于药用植物转录组分析中。本研究首次采用高通量测序技术对玄参根进行转录组测序和功能分析,深一步挖掘其次生代谢相关基因,填补了玄参转录组信息的空白。测序数据采用Trinity软件共拼接得到73 983条unigene,平均序列长度823 bp,约73%的reads参与了拼接,拼接的N50长度为1 546 bp,所测得的unigene数量基本涵盖了全部转录组信息。测序数据质控合格,测序质量良好。获得如此大的序列信息量,表明高通量测序技术是批量发现玄参功能基因的有效手段。本研究利用生物信息学方法对拼接序列进行注释和功能分类,其中56 389条unigene在Blast、同源性搜索中得到注释,注释率达76.2%,剩下的17 594条unigene可能是由于长度较短而未与公共数据库中的序列比对上,也可能是非编码序列或者是新的基因[21]。
本研究通过同源搜索,共发现520条编码玄参次生代谢途径关键酶的相关基因和191个相关转录因子。这些基因的发现,为后续开展的玄参次生代谢物合成关键基因的鉴定和克隆提供了基础数据。众所周知,萜类物质结构复杂,化学合成较困难,目前主要以原植物提取获得。因此,开展玄参次生代谢物合成关键酶基因及转录因子的表达调控分子机制尤为重要,随着后基因组工作的深入,这些关键基因将作为改造植物代谢途径的有力工具,人为控制次生代谢物的合成量。本研究所获得的转录组信息不光为玄参次生代谢物生物合成研究提供基础数据,同时也为进一步开展玄参生长发育、抗病抗逆等相关分子机制研究提供可靠信息。
此外,与传统测序方法相比,高通量测序技术操作简单,能够挖掘出大量的SSR位点信息。本研究发现玄参根SSR位点11 659个,重复类型以二核苷酸为主,占全部SSR的40.13%。这些SSR位点的发现可为玄参分子标记的开发、群体遗传多样性分析、种质鉴定、标记辅助选择、基因定位、亲缘鉴定等方面的研究提供依据。
由于玄参未开展全基因组测序,可供参考的遗传信息非常少,因此对玄参根转录组的特性分析还有待于进一步的深入研究。本研究所获得的玄参根转录组信息,一方面获得大量SSR位点,为后续研究玄参的功能基因克隆及遗传多样性研究提供参考;另一方面获得了丰富的参与次生代谢的关键基因,也为玄参药用成分的生物合成和调控机制奠定基础。
[参考文献]
[1] 中国药典.一部[S].2015:108.
[2] Qian J, Hunkler D, Safayhi H, et al. New iridoid-related constituents and the anti-inflammatory activity of Scrophularia ningpoensis[J]. Planta Med, 1991, 57: 56.
[3] Grabherr M G, Haas B J, Yassour M, et al..Full-length transcriptome assembly from RNA-Seq data without a reference genome[J]. Nat Biotechnol, 2011, 29, 644.
[4] Fullwood M J, Wei C L, Liu E T,et al. Next-generation DNAsequencing of paired-end tags for transcriptom and genome analysis[J]. Genome Res, 2009, 19(4): 521.
[5] Dassanayake M,Haas J S,Bohnert H J,et al. Shedding light on an extremophile life style through transcriptomics[J]. New Phytologist,2010, 183 (3): 764.
[6] Lu T T,Lu G J,Fan D L,et al. Function annotation of the rice transcriptome at single-nucleotide resolution by RNA-Seq[J]. Genome Res,2010, 20 (1):1238.
[7] Sangwan R S,Tripathi S,Singh J,et al. De novo sequencing and assembly of Centella asiatica leaf transcriptome for mapping of structural,functional and regulatory genes with special reference to secondary metabolism[J]. Gene,2013, 525 (2): 58.
[8] Grabherr M G, Haas B J, Yassour M, et al.Full-length transcriptome assembly from RNA-Seq data without a reference genome[J]. Nat Biotechnol, 2011, 29: 644.
[9] Conesa A, Gtz S, García-Gómez J M, et al. Blast GO: a universal tool for annotation, visualization and analysis in functional genomics research[J]. Bioinformatics, 2005, 21(18):3674.
[10] Altschul S F, Madden T L, Schffer A A, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs[J]. Nucleic Acids Res, 1997, 25:3389.
[11] Young M D, Wakefield M J, Smyth G K, et al. Gene ontology analysis for RNA-seq: accounting for selection bias[J]. Genome Biol, 2010, 11 (2) :R14.
[12] Gtz S, García-Gómez J M, Terol J, et al. High-throughput functional annotation and data mining with the Blast2 GO suite[J]. Nucleic Acids Res, 2008, 36: 3420.
[13] Kanehisa M, Araki M, Goto S, et al. KEGG for linking genomes to life and the environment[J]. Nucleic Acids Res, 2008, 36:480.
[14] Simbaqueba J, Sanchez P, Sanchez E, et al. Development and characterization of microsatellite markers for the cape gooseberry physalisperuviana[J]. PLoS ONE, 2011, 6(10): e26719.
[15] 趙恒伟,葛峰,孙颖,等. 植物萜类物质生物合成的相关转录因子及其应用前景[J].中草药,2012,10(43):2512.
[16] Iwase A, Mitsuda N, Koyama T, et al. The AP2/ERF transcription factor WIND1 controls cell dedifferentiation in Arabidopsis [J]. Curr Biol, 2011, 21(6): 508.
[17] Qi W W, Sun F, Wang Q J, et al. Rice ethylene-response AP2/ERF factor OsEATB restricts internode elongation by down-regulating a gibberellin biosynthetic gene [J]. Plant Physiol, 2011, 157(1): 216.
[18] Yang C Y, Hu F C, Li J P, et al. The AP2/ERF transcription factor AtERF73/HRE1 modulates ethylene responses during hypoxia in Arabidopsis [J]. Plant Physiol, 2011, 156(1): 202.
[19] Rushton P J, Somssich I E, Ringler P, et al. WRKY transcription factors [J]. Trends Plant Sci, 2010, 15(5): 1360.
[20] Tripathi P, Rabara R C, Langum T J, et al. The WRKY transcription factor family in Brachypodium distachyon[J]. BMC Genomics, 2012, 13(270): 1.
[21] 李滢,孙超,罗红梅,等. 基于高通量测序 454 GS FLX的丹参转录组学研究[J]. 药学学报,2010,45(4):524.
[责任编辑 吕冬梅]
猜你喜欢
大众科技(2022年10期)2022-11-11
云南化工(2021年7期)2021-12-21
中成药(2018年6期)2018-07-11
中成药(2016年4期)2016-05-17
