
磁性材料磁有序的分子场来源∗
2017-08-03 08:10齐伟华李壮志马丽唐贵德吴光恒胡凤霞
物理学报 2017年6期
齐伟华 李壮志 马丽 唐贵德†吴光恒 胡凤霞
1)(河北师范大学物理科学与信息工程学院,河北省新型薄膜材料实验室,石家庄 050024)
2)(中国科学院物理研究所磁学国家重点实验室,北京 100190)
(2016年11月15日收到;2016年12月8日收到修改稿)
磁性材料磁有序的分子场来源∗
齐伟华1)李壮志1)马丽1)2)唐贵德1)2)†吴光恒2)胡凤霞2)
1)(河北师范大学物理科学与信息工程学院,河北省新型薄膜材料实验室,石家庄 050024)
2)(中国科学院物理研究所磁学国家重点实验室,北京 100190)
(2016年11月15日收到;2016年12月8日收到修改稿)
对于磁性材料磁有序能的来源,即外斯分子场来源,本文提出一个模型:在磁性金属和合金中的相邻离子实之间,以及磁性氧化物的相邻阴阳离子间,其外层轨道上高速运动的电子分别有一定概率形成三种不同的状态.1)具有一定寿命的自旋相反的电子对,称为外斯电子对(WEP);2)距离很近且自旋方向相同的电子,容易发生互相交换,交换前后电子的自旋方向保持不变;3)当一个离子外层轨道有2个电子,其相邻的离子外层轨道只有1个电子时,前者多出的电子可以跃迁到后者的轨道上,并且保持自旋方向不变.我们认为,W EP两个电子间的静磁吸引能是分子场(即磁有序能)的主要来源.进而,推导出WEP的能量表达式、两电子的平衡间距和最大间距,探讨了在几种钙钛矿结构锰氧化物中形成外斯电子对的概率,用以解释居里温度附近晶格常数随温度变化的特点.结果表明这个模型是合理的.
磁有序模型,磁性氧化物,磁性金属和合金
1 引 言
1.1 磁畴和外斯分子场
铁磁性、亚铁磁性和反铁磁性材料中存在磁畴.利用偏光显微镜可在石榴石磁泡薄膜材料上观察到十分清晰的磁畴[1,2],如图1所示.其中磁畴的宽度在微米数量级,与薄膜的厚度有关.膜越薄,磁畴越窄.在图1中的磁畴存在N,S极,黑白磁畴代表相反的磁化方向.图1(a)是无外加磁场时的一种磁畴.图1(b)是经过一个直流磁场和一个脉冲磁场联合作用,并且脉冲磁场撤销后的情况,其中外磁场的方向与黑畴磁化方向平行.这说明在一个磁畴中原子磁矩克服巨大的磁性排斥能有序排列起来.为了解释使原子磁矩有序排列的巨大能量,1907年,外斯提出了分子场假说[3].按照这个假说:铁磁性物质中包含有许多小区域,即使没有外磁场,这些区域中也有自发磁化强度.这些具有自发磁化强度的小区域称为磁畴.磁畴内的自发磁化是由于晶体中有很强的内场而产生的,这种内场可称为分子场[3].

图1 石榴石磁泡薄膜材料的两种磁畴[1,2]Fig.1. M agnetic dom ains of the garnet bubblefi lm[1,2].
戴道生和钱昆明[3]在介绍分子场理论时,估算了分子场的数值:当温度达到居里温度时,自发磁化消失,此时原子的热运动能量与自发磁化的能量相当,即
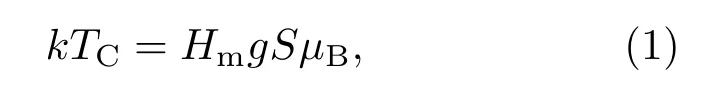
其中Hm代表分子场强度,玻尔磁子µB=9.271× 10−21erg/Oe(1 erg/Oe=10−3J/T).对于金属Fe,居里温度TC=1043 K,当平均每个原子的磁矩近似为gSµB=2.22µB时,可计算出Hm= 6.994×106Oe(1 Oe=79.5775 A/m).
对于钙钛矿结构锰氧化物La0.8Ca0.2M nO3, La0.75Ca0.25M nO3和La0.70Sr0.30M nO3,令(1)式中的gS等于每个分子的平均磁矩µobs,可计算出这些样品的分子场强度Hm.计算结果和计算过程所用到的参数列于表1(1 emu/cm3=103A/m).对于如此巨大的分子场的来源,至今没有得到满意的解释,成为长期困扰铁磁性物理研究者的难题.这可能是多年来磁学研究难以取得突破性进展的主要原因之一.所以探讨这个问题具有十分重要的意义.
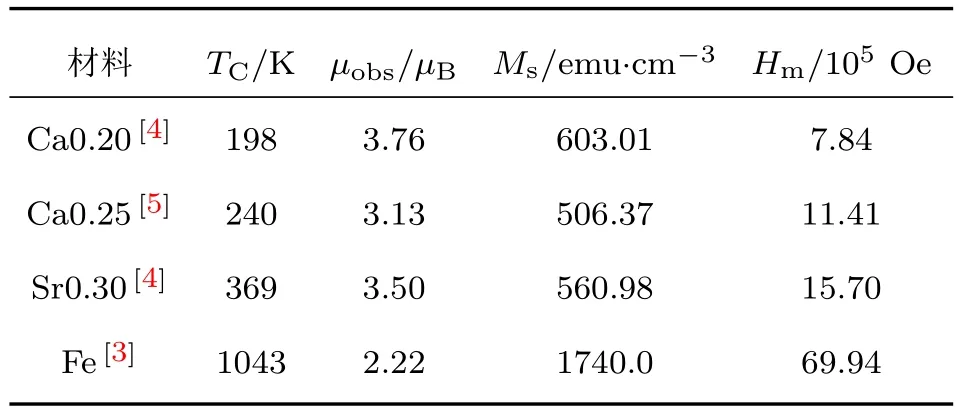
表1 La0.8 Ca0.2M nO 3,La0.75Ca0.25M nO 3,La0.70Sr0.30-M nO3和金属铁的居里温度TC、平均分子磁矩实验值µobs、饱和磁化强度Ms和分子场强度HmTab le 1.Curie Tem peratu re TC,averagem olecu lar m agnetic m om entµobs,satu ration m agnetization Msand the m olecular field intensity Hmof La0.8Ca0.2M nO3, La0.75Ca0.25M nO 3,La0.70 Sr0.30M nO 3 and m etal iron.
1.2 铁磁性物理学领域的几个长期存在争议的问题
到目前为止,磁学界普遍用原子间电子的交换作用解释分子场的来源[3,4−10].称金属和合金磁性材料中的交换作用为直接交换作用,称氧化物铁磁材料中磁性原子间的反铁磁耦合为超交换作用,铁磁耦合为双交换作用.此外,对于一些氧化物的磁有序问题,用晶体场理论配合超交换和双交换作用给出解释;对于金属和合金磁有序问题,用固体能带论给出解释.然而,对于金属磁性和氧化物磁性之间的关系,没有发现满意的解释.即使对于涉及到氧化物磁有序的超交换和双交换作用之间的联系,也没有发现满意的解释.特别是,对于材料磁性的一些实验结果,利用现有的模型还不能给出合理的解释,因而存在着一些长期困扰磁学界的问题,举例如下.
问题1 对于(A)[B]2O4型尖晶石结构铁氧体M Fe2O4(M=Fe,Co,Ni,Cu),按照传统理论,其(A),[B]子晶格中离子磁矩分别平行排列,但两个子晶格的离子磁矩相互反平行排列.可近似认为其中Fe离子都是3价,一半进入(A)位,另一半进入[B]位,所以Fe离子的磁矩恰好相互抵消;M离子都是2价,并且全部进入[B]位,所以当M=Fe, Co,Ni,Cu时,其平均每个分子的磁矩实验值分别为4.2,3.3,2.3,1.3µB,略大于二价Fe,Co,Ni, Cu离子的磁矩4,3,2,1µB.这种差别指出,实际上一部分二价M和Fe离子进入了(A)位.当M=M n时,实验磁矩约为4.6µB,略小于M n2+的磁矩5µB.当M=Cr时,实验磁矩约为2µB,只有Cr2+磁矩4µB的二分之一.如图2所示.迄今为止,对于M n和Cr掺杂尖晶石铁氧体的磁结构与Fe,Co,Ni和Cu掺杂时的区别,仍存在广泛的争议[11−25],成为长期困扰磁学界的一个问题,以至于在经典的铁磁性物理著作中[3,9,10],回避关于Cr掺杂尖晶石铁氧体问题的介绍.
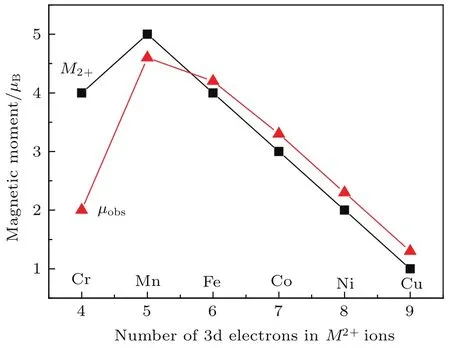
图2 (A)[B]2O4型尖晶石结构铁氧体M Fe2O4的分子磁矩实验值µobs和二价离子磁矩M2+随M2+离子中3d电子数目的变化Fig.2.Dependences on the number of 3d electrons of the experim ental m olecu lar m agnetic m om ent,µobs, and d ivalent M2+ion m agnetic m om ent,M2+,for M Fe2O4ferrites w ith(A)[B]2O4sp inel structure.
问题2 对于典型的ABO3型钙钛矿结构锰氧化物La1−xSrxMnO3,当x>0时,样品的磁矩逐渐增大;当x达到0.15时,样品分子磁矩的实验值最大,为4.2µB[8].传统观点认为在这种材料中,Mn3+—O2−—Mn3+离子链间由于超交换作用而导致反铁磁耦合,M n3+—O2−—M n4+离子链间因双交换作用而导致铁磁耦合.然而,当x达到0.15时,即使Mn3+和Mn4+离子全部平行排列,其分子磁矩的计算值也只有3.85µB,明显小于实验值4.2µB.
问题3 对于典型磁性金属Fe,Co,Ni,其平均原子磁矩实验结果分别为2.22,1.72,0.62µB[3], 0◦C时的电阻率分别为8.6,5.57,6.14µΩ·cm[26].铁磁性物理学利用固体能带论解释其平均原子磁矩的实验结果[3,9,10],但是没有发现关于其电阻率与这些磁矩实验值之间关系的满意解释.此外,金属和合金中原子的平均磁矩显著小于氧化物中金属离子的磁矩,对于其原因,也存在争议.
问题4 一些典型金属和氧化物磁性材料的晶体结构、平均分子磁矩和居里温度列于表2[4,5,8,26].可以看出,尖晶石结构铁氧体的居里温度与金属Ni比较接近;而钙钛矿结构锰氧化物的居里温度显著降低.这说明传统观点中解释金属磁性的直接交换作用与解释氧化物磁性的超交换和双交换作用之间必然存在本征的联系.而对于这种本征的联系,没有发现满意的解释.
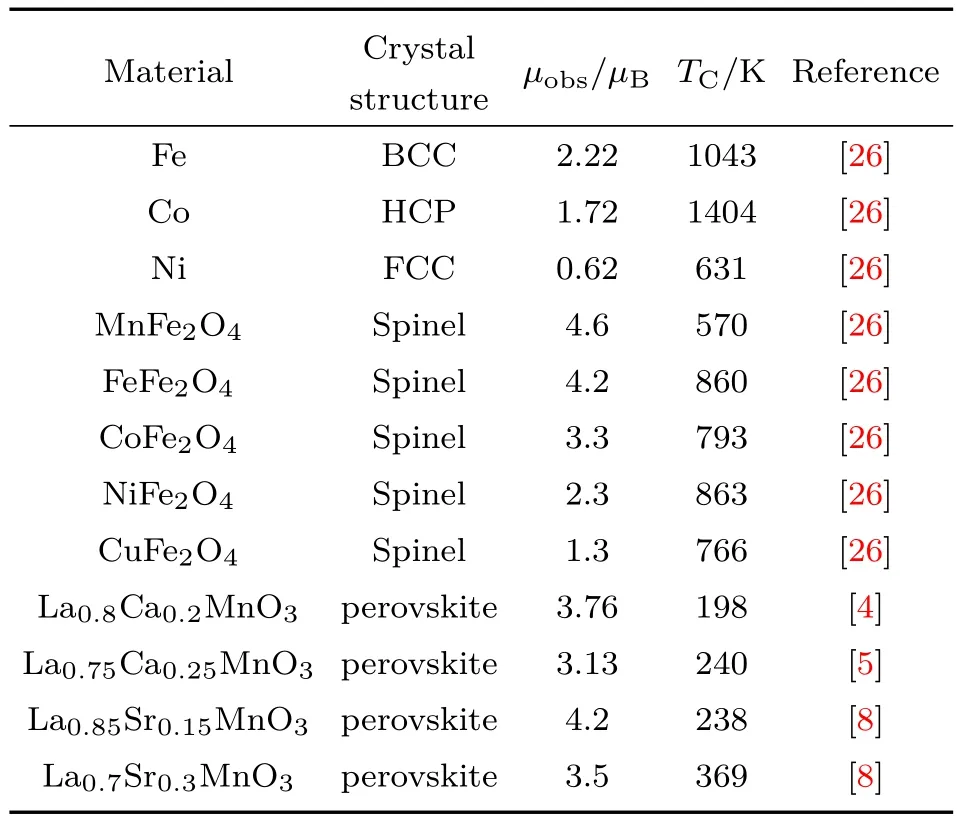
表2 一些典型金属和氧化物磁性材料的晶体结构、平均分子磁矩µobs和居里温度TCTab le 2.Crystal structu re,average m olecu lar m agnetic m om entsµobsand Curie tem peratu re TCof severalm etals and oxides.
1.3 近几年本课题组对于上述问题1—3的一些有益探讨
首先,我们提出一个O 2p巡游电子模型,不仅可以替代超交换和双交换作用模型解释氧化物的磁有序问题,而且可解释长期困扰磁学界的上述问题1和问题2[27−35].这个模型包括三点:1)考虑到磁性氧化物的电离度[36,37],其中除O2−离子外还存在一部分O1−离子[38−42],在O1−离子外层电子轨道存在1个O 2p空穴.因此O2−离子外层轨道的2p电子就有一定的概率以阳离子为媒介跃迁到相邻O1−离子的2p空穴上,并保持自旋方向不变,形成巡游电子;2)一个O2−离子外层轨道上自旋方向相反的两个O 2p电子分别成为A,B子晶格中的巡游电子;3)在每个子晶格中,O 2p巡游电子在跃迁过程中保持自旋方向不变,阳离子的3d电子(包括局域电子和巡游电子)自旋的方向必须遵守洪特定则;又因为3d过渡族金属的3d壳层最多只能容纳5个自旋方向相同的电子[43],如果两个阳离子的3d电子数目都满足nd≤4或nd≥5,这两个离子的磁矩方向相同;如果一个阳离子的nd≤4,另一个阳离子的nd≥5,这两个离子的磁矩方向相反.
其次,我们提出一个关于金属磁性的新型巡游电子模型,成功地解释了上述问题3[44].这个模型包括:1)在3d过渡族原子结合成金属的过程中,由于受到原子间电子泡利排斥力的挤压作用,原子的大部分4s电子进入3d轨道,变成3d电子,剩余的4s电子作为自由电子,为讨论方便,我们把金属中的离子实简称为离子,其中不含自由电子;2)由于进入3d轨道的4s电子数目平均值不是整数,一部分离子就会比另一部分离子多出一个3d电子,这些多出的3d电子在邻近原子间发生跃迁,形成巡游电子,巡游电子在跃迁过程中保持自旋方向不变,除自由电子和巡游电子外,剩余的3d电子都是局域电子;3)金属的电阻率随自由电子含量的增加而减小.自由电子的迁移过程与电子的自旋方向无关,自由电子的自旋对材料磁矩也没有贡献,金属中巡游电子跃迁的概率远小于自由电子.所以,与自由电子相比,巡游电子跃迁对金属电阻率的贡献较小.根据这个模型,我们利用Fe,Ni,Co磁矩的实验值计算出其中的自由电子含量,发现Fe,Ni, Co,Cu的电阻率随其中自由电子浓度的增加而减小,从而首次成功解释了金属磁性与电阻率之间的关系.
本文对上述问题4进行探讨,研究金属磁性与氧化物磁性之间的本征联系,即外斯分子场的来源.
2 关于磁有序能的外斯电子对(WEP)模型及其应用举例
2.1 W EP模型
对比上述O 2p巡游电子模型和金属磁性的新型巡游电子模型可以看出,其中的巡游电子都是在相邻离子的外层轨道间跃迁,这种跃迁必然受到离子外层轨道对于电子自旋方向的限制.因此,我们所称的巡游电子不包含金属中的自由电子,即磁性金属中的电子分为自由电子、巡游电子和局域电子;而磁性氧化物中只含有巡游电子和局域电子.
根据Shannon[45]对有效离子半径的研究,离子的化合价每变化一价,其有效半径都存在明显差别.这说明单晶体和多晶体中离子的外层电子轨道可理解为半径在一定范围内的电子云壳层.表3列出了几种离子的二、三价有效半径及其半径差.可见这些离子的二、三价半径差在0.09—0.19Å之间.从这个角度看来,在上述巡游电子模型中,无论是O 2p巡游电子模型中电子在相邻的阴阳离子间跃迁,还是磁性金属中巡游电子在相邻的金属离子间跃迁,都具有相似之处.
表3 几种阳离子在配位数为6时的二、三价有效半径,及其半径差−[45]Tab le 3.Divalent and trivalent eff ective radii,,,and their d iff erence−of several cations w ith coord ination num ber 6[45].
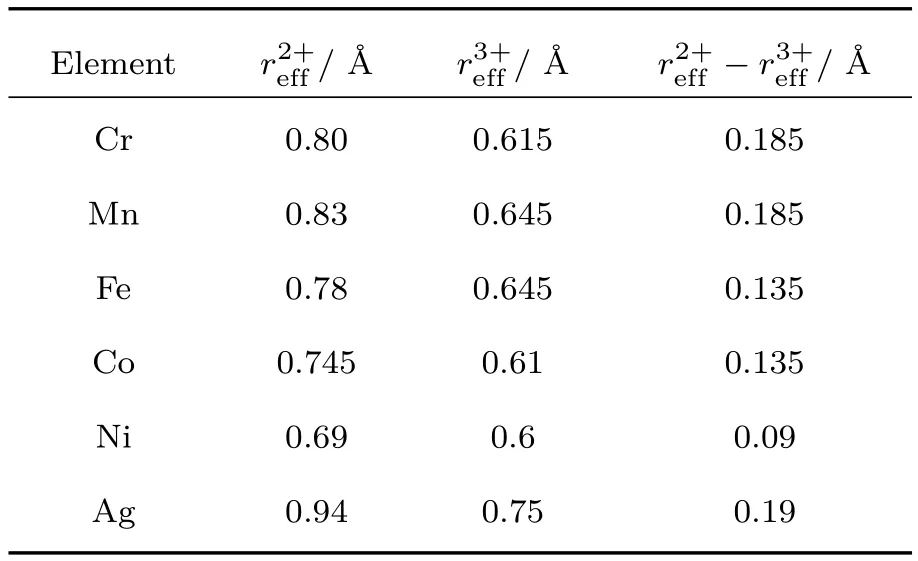
表3 几种阳离子在配位数为6时的二、三价有效半径,及其半径差−[45]Tab le 3.Divalent and trivalent eff ective radii,,,and their d iff erence−of several cations w ith coord ination num ber 6[45].
注:在配位数为6时O2−离子的有效半径为1.40Å[45]. Note:Eff ective radius of O2−w ith coordination number 6 is 1.40Å[45].
E lem ent r2+eff/Å r3+eff/Å r2+eff−r3+eff/Å Cr 0.80 0.615 0.185 M n 0.83 0.645 0.185 Fe 0.78 0.645 0.135 Co 0.745 0.61 0.135 N i 0.69 0.6 0.09 Ag 0.94 0.75 0.19
假设电子在一个离子外壳层中高速运动时自旋方向不变,由于相邻离子的最外层轨道十分接近,其电子分别有一定概率处于图3(a)—(c)所示的状态.1)当两离子处于图3(a)的状态时,离子间的两电子自旋磁矩反平行(称为WEP),产生静磁吸引能,同时也存在泡利排斥能,从而可处于吸引能和排斥能的短暂平衡态,具有确定的平衡间距和寿命,这种情况下,电子不能在两离子间交换; 2)当两离子处于图3(b)所示状态时,两电子的自旋磁矩平行,容易发生互相交换,交换前后电子的自旋方向保持不变,并且这种状态的两个电子间在交换前存在磁性排斥能;3)当一个离子外层轨道有两个电子,其相邻的离子外层轨道只有一个电子,且处于图3(c)的状态时,左侧离子上的电子可以跃迁到右侧离子上,并且保持自旋方向不变.这就是在磁性材料中巡游电子的自旋方向保持不变的原因,如果自旋方向不同,就不能发生图3(b)所示的交换或图3(c)所示的巡游.我们把图3(b)和图3(c)所示的跃迁统称为巡游电子的跃迁.
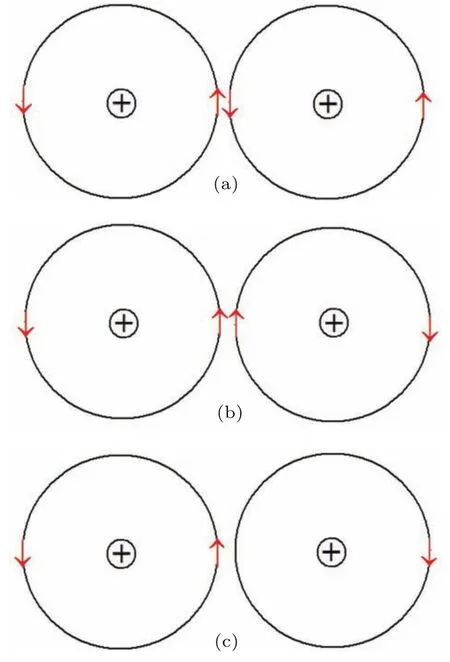
图3 近邻离子外层电子轨道的(a)W EP和(b),(c)巡游电子示意图Fig.3.Illustration of(a)W EP and(b),(c)itinerant electron between ou ter orbits of the ad jacent ions.
2.2 几种典型材料的外斯分子场能量估算
以Fe为例,根据文献[3],Fe饱和磁化强度Ms为1740 Gs(1 Gs=10−4T).根据表1,分子场强度Hm为6.994×106Oe,则分子场的能量密度(1 erg/cm3=10−1J/m3)可以计算如下:
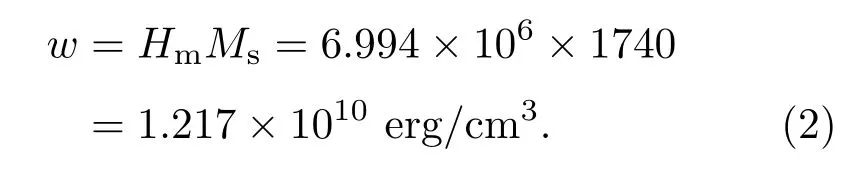
铁具有体心立方晶格,晶格常数2.86Å,每个晶胞中含2个Fe原子,则平均每对Fe原子的分子场能量为
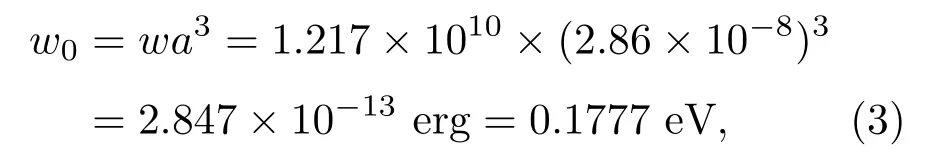
它形成一个使晶格体积收缩的力. 与平均每对离子的结合能(约10 eV)比较,这个值是合理的.对于钙钛矿结构锰氧化物La0.8Ca0.2MnO3, La0.75Ca0.25MnO3和La0.70Sr0.30MnO3,利用表1中的参数,同样可算出分子场的能量密度w.对于平均每对Mn离子间的分子场能量w0,计算时注意到每个分子含一个M n离子;La0.8Ca0.2M nO3和La0.75Ca0.25MnO3为正交结构,每个晶胞中有4个分子,w0=wv/2;La0.70Sr0.30MnO3为菱面体结构,每个晶胞中有6个分子,w0=wv/3.其中v为晶胞体积.计算结果列于表4.
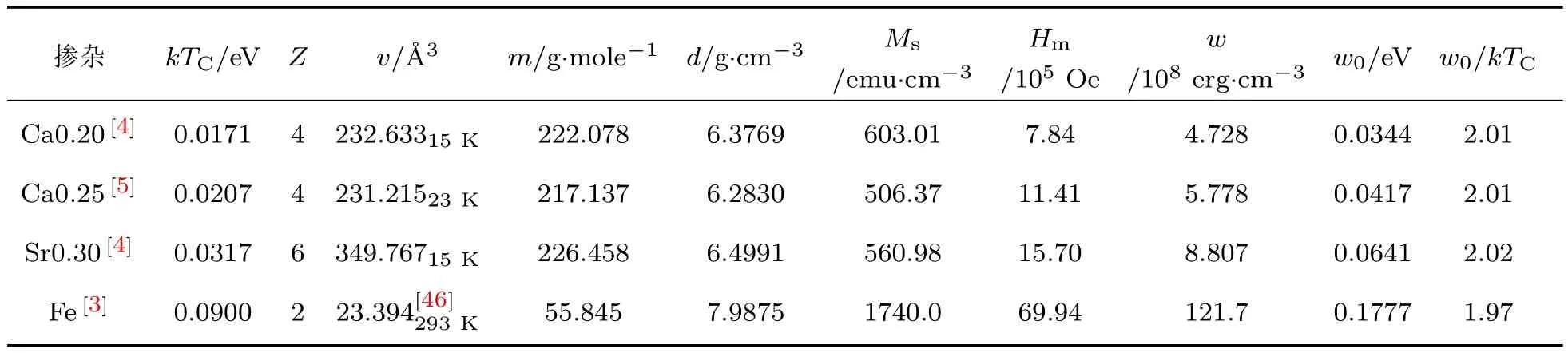
表4 La0.8Ca0.2M nO3,La0.75Ca0.25M nO3,La0.70Sr0.30M nO3和金属铁中每对磁性离子间的外斯分子场能w0以及相关的参数,其中kTC是在居里温度TC处的热能,Z是每个晶胞中的分子数,v和m分别是晶胞的体积和质量,d是材料的密度,Ms和Hm分别是饱和磁化强度和外斯分子场强度Tab le 4.Weiss m olecu lar field energy,w0,per pair m agnetic ions,and relative param eters of La0.8Ca0.2M nO3, La0.75Ca0.25M nO3,La0.70Sr0.30M nO3and m etal iron,w here kTCis therm al energy at Cu rie Tem perature TC,Z is them olecu le num ber per crystal cell,v and m are the volum e and m ass of a crystal cell,d is the density,Msand Hmare the saturation m agnetization and W eissm olecu lar field intensity.
从表4看到一个非常有趣的结果:对于La0.8-Ca0.2MnO3,La0.75Ca0.25MnO3,La0.70Sr0.30MnO3和金属Fe,尽管其结构不同,磁性离子间距不同,其平均每对磁性离子间的分子场能量与居里温度相应热能(kTC)的比值分别为2.01,2.01,2.02和1.97,这4个比值十分接近.说明我们关于这几种材料分子场能量的计算方法是合理的.
2.3 居里温度附近的热膨胀现象
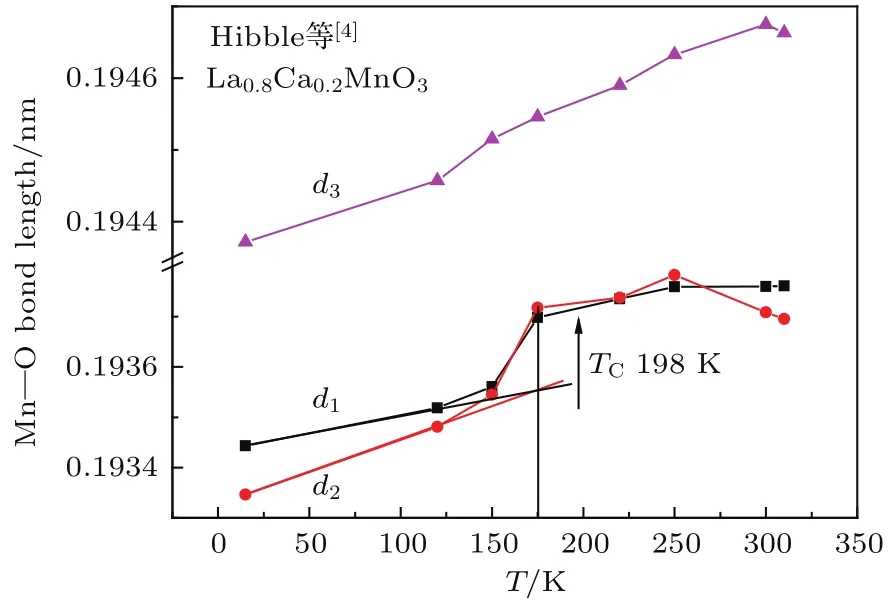
图4 (网刊彩色)正交结构的La0.8Ca0.2M nO3样品中M n—O离子间距d1,d2,d3随温度的变化[4]Fig.4.(color on line)Dependences on tem peratu re of the distances between M n and O,d1,d2,d3,for orthorhombic La0.8Ca0.2M nO3sam p le[4].
Hibble等[4]研究了正交结构的La0.8Ca0.2-M nO3样品晶格常数随温度的变化,从而可估算出沿三个相互垂直的方向上Mn—O离子间距d1, d2,d3随温度的变化情况,结果如图4所示.可以看到,在居里温度附近d1,d2有一个迅速增大的过程,而d3的变化较小.由此我们推测Mn离子的磁矩沿d3晶轴方向排列,WEP的静磁作用力主要沿d1,d2晶轴,这是一个使离子间距收缩的力.当温度接近并逐渐达到居里温度附近时,WEP的静磁作用力迅速减小,导致Mn—O离子间距迅速增大.因此,我们对d1,d2低温段做切线,近似认为低温段的线性变化与磁有序能无关,从120 K到175 K过程中d1,d2的测量值偏离切线是由于WEP的静磁能逐渐减小,最后消失引起的.由此估算出在175 K时d1,d2的测量值与切线处的差Δdobs为0.00147和0.00167Å.Radaelli等[5]研究了正交结构的La0.75Ca0.25MnO3样品晶格常数随温度的变化,从而可计算出沿三个相互垂直的方向上Mn—O离子间距d1,d2,d3随温度变化的情况,结果如图5所示.Hibble等[4]研究了菱面体结构La0.7Sr0.3M nO3样品的晶格常数随温度的变化,其d2与d1相等,d1和d3随温度的变化示于图6,其居里温度为369 K[8].与图4类似,可估算出Ca掺杂0.25和Sr掺杂0.3样品中由于WEP的静磁能消失引起的M n—O间距变化量Δdobs,列于表5.可见Ca掺杂0.2,Ca掺杂0.25和Sr掺杂0.3这三个样品的Δdobs值依次增大.这是因为3个样品的居里温度依次升高,分别为198,240和369 K.
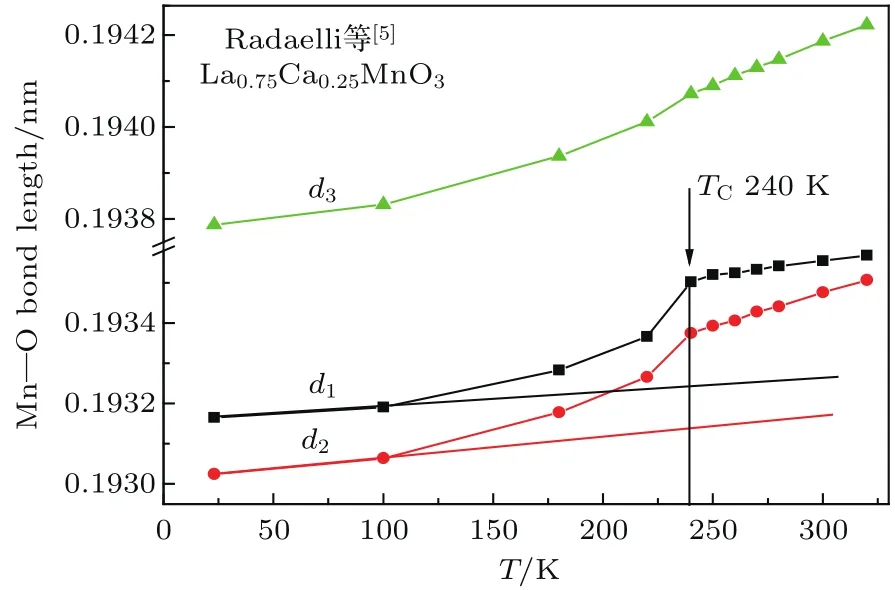
图5 (网刊彩色)正交结构的La0.75Ca0.25M nO3样品中M n—O离子间距d1,d2,d3随温度的变化[5]Fig.5.(color on line)Dependences on tem peratu re of the distances between M n and O,d1,d2,d3,for orthorhom bic La0.75Ca0.25M nO3sam p le[5].
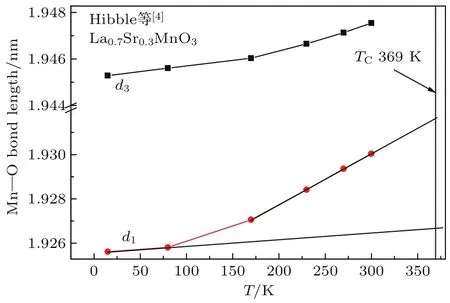
图6 (网刊彩色)菱面体结构的La0.7Sr0.3M nO3样品中M n—O离子间距d1,d3随温度的变化[4]Fig.6.(color on line)Dependences on tem peratu re of the distances between M n and O,d1and d3,for rhombohed ral La0.7Sr0.3M nO3sam p le[4].
2.4 W EP能量及其与材料热膨胀的关系
设图3(a)所示WEP两电子间的平均距离为re,随着re的减小,电子间的泡利排斥能迅速增大.假设两个电子处于图3(a)所示状态的概率为D,各自带有电荷−e,自旋磁矩1µB,由此导致系统的能量增量,即磁有序能为
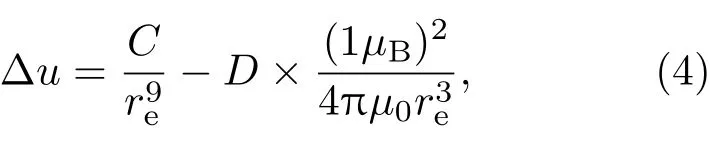

设Δre=rem−re0代表WEP两电子最大间距与平衡间距之差.当WEP两电子间距变化小于Δre时,两电子间的静磁吸引能倾向于使电子间距减小,可维持WEP的存在.随着温度的升高,当WEP两电子间距变化大于Δre时,两电子间的静磁吸引能不再使电子间距减小,WEP消失,分子场消失.对于La0.8Ca0.2MnO3,La0.75Ca0.25MnO3和La0.70Sr0.30MnO3,令(10)式中的|Δu0|等于前述计算出的平均每对M n离子间的分子场能量w0,通过调整参数D,使Δre等于在居里温度附近由于磁有序能消失造成M n—O键长d1,d2变化量的测量值Δdobs,相关的参数列于表5.可见,参数D的值在0.07%到3.13%之间,说明图3(a)所示WEP只要有0.07%—3.13%的形成概率,所产生的磁有序能就可使这三个样品产生相应的自发磁化.此外,对于这三个样品,WEP电子间最大间距rem小于0.035Å,明显小于表3中给出的外电子壳层厚度的最小值0.09Å.这些结果说明我们关于WEP电子间的静磁能是磁有序能主要来源的模型至少在定性上是合理的.

表5 对于La0.8Ca0.2M nO3,La0.75Ca0.25M nO3和La0.7Sr0.3M nO3,W EP形成概率D和相关的参数.w0是平均每对M n离子间的分子场能量;Δdobs是在居里温度附近由于磁有序能消失造成M n—O键长d1,d2的变化量的测量值;re0和rem分别是W EP平衡间距和最大间距;Δre=rem−re0Tab le 5.Probability of form ed W EP,D,and relative param eters of La0.8Ca0.2M nO3,La0.75Ca0.25M nO3, La0.70Sr0.30M nO3.w0ism olecu lar field energy per pair m agnetic ions.Δdobsis the variation of M n—O bond length near the Curie tem perature.re0and remare the balance d istance and m axim um d istance between electrons of W eiss electron pair,respectively.Δre=rem−re0.
2.5 磁有序能的其他主要影响因素
表5中三个样品的参数D在0.07%—3.13%之间,存在数量级的差别,说明除WEP间的静磁能之外还存在影响磁有序能的其他因素.实际上,当两个相邻离子间外层轨道电子处于图3(b)所示状态时,存在磁性排斥力.此外在一个磁畴中磁性离子的铁磁性耦合导致离子磁矩间存在排斥力,后一种排斥力可以等效为如图7所示的电子自旋之间的排斥力.这两种排斥力也与电子之间距离的三次方成反比.因而,(8)式可改写为
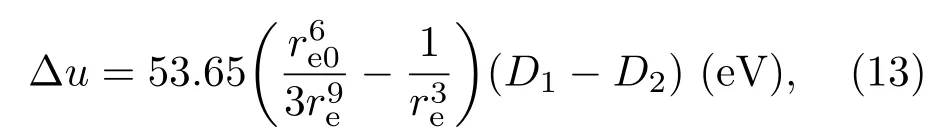
其中D1代表WEP电子间的静磁吸引能系数,它只与外层轨道的电子态有关;D2代表离子间的磁性排斥能系数,它不仅与外层轨道电子状态有关,而且与外层轨道所在的次壳层(例如3d电子壳层)电子数目有关.
此外,对于(A)[B]2O4型尖晶石结构铁氧体M Fe2O4(M=Fe,Co,Ni,Cu),用任何元素替代Fe,使Fe含量小于2,将导致居里温度迅速下降.对于ABO3型钙钛矿结构锰氧化物La1−xSrxM nO3,用任何其他过渡金属元素替代Mn,也会导致居里温度迅速下降.前者中存在大量Fe3+离子,后者中存在大量M n3+离子,分别有5个和4个3d电子.根据本课题组提出的O 2p巡游电子模型,当O 2p电子以Fe3+离子或Mn3+离子为媒介在氧离子间巡游时,都占据3d次电子壳层的最高能级,当O 2p电子以其他离子为媒介在氧离子间巡游时,将占据3d次电子壳层的较低能级.这说明只有相邻离子的最外层电子都属于外壳层的最高能级时,形成WEP的概率才可能比较大;否则,形成WEP的概率大幅减小,导致磁性吸引能减小.此外,如果巡游电子的巡游过程中需要在不同的能级间跃迁,相对于只在最高能级间跃迁消耗的能量比较多.用D3表示这些因素的影响,上述(13)式可修改为
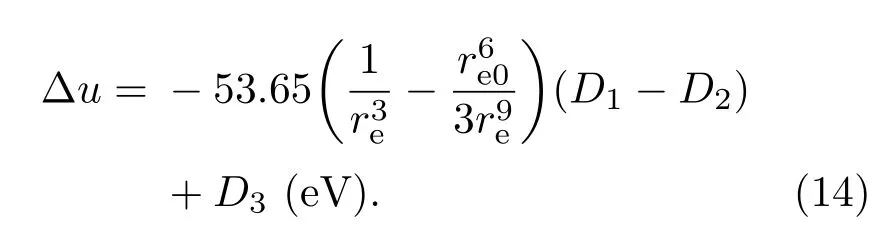
关于(13)和(14)式所涉及的问题,还有待于结合相应的实验数据进行深入研究.
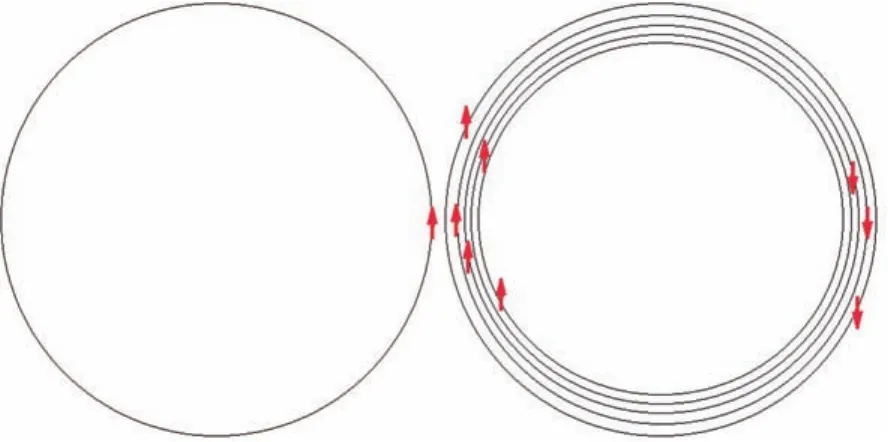
图7 相邻离子间电子自旋排斥力示意图Fig.7.Illustration of repelling force between electron sp ins for the ad jacent ions.
3 结 论
本文首次提出了一个自旋相反的外斯电子对模型,即WEP模型,用于解释外斯分子场的主要来源.WEP的吸引能来自相邻离子自旋相反的电子磁矩间的静磁吸引能,排斥能来自电子间的泡利排斥能,WEP具有一定的形成概率,也可等效为具有一定寿命.对于钙钛矿结构锰氧化物La0.8Ca0.2MnO3,La0.75Ca0.25MnO3和La0.7Sr0.3M nO3,如果不考虑其他因素,在相邻离子间电子绕离子实运动过程中形成这种WEP的概率参数D分别只要有0.07%,0.31%和3.13%,就可造成材料的自发磁化,并具有相应的居里温度.关于这三个样品参数D存在数量级的差别,是因为磁有序能还受到电子自旋磁矩排斥能等因素的影响.本文提出的WEP模型反映了O 2p巡游电子模型和金属中新型巡游电子模型之间的本征联系.这为发展系统的铁磁性物理基础理论提供了新的思路.
[1]Han B S,N ie X F,Tang G D,X i W 1985 Acta Phys. Sin.34 1396(in Chinese)[韩宝善,聂向富,唐贵德,奚卫1985物理学报34 1396]
[2]Tang G D,M a C S,Yang L X,M a L M 2003 M odern Physics Experim ents(Shijiazhuang:Hebei Science and Technology Press)p148(in Chinese)[唐贵德,马长山,杨连祥,马丽梅 2003近代物理实验 (石家庄:河北科学技术出版社)第148页]
[3]Dai D S,Qian K M 1987 Ferrom agnetism(Beijing:Science Press)p103(in Chinese)[戴道生,钱昆明1987铁磁学(上册)(北京:科学出版社)第103页]
[4]H ibb le S J,Cooper S P,Hannon A C,Faw cett I D, G reenblatt M 1999 J.Phys.:Condens.M atter 11 9221
[5]Radaelli P G,Cox D E,M arezio M,Cheong SW,Shiff er P E,Ram irez A P 1995 Phys.Rev.Lett.75 4488
[6]Schiff er P,Ram irez A P,BaoW,Cheong SW 1995 Phys. Rev.Lett.75 3336
[7]M ahend iran R,T iwary S K,Raychaudhu ri A K,Ram akrishnan T V 1996 Phys.Rev.B 53 3348
[8]U rushibara A,M oritom o Y,A rim a T,Asam itsu A,K ido G,Tokura Y 1995 Phys.Rev.B 51 14103
[9]Chikazum i S 1997 Physics of Ferrom agnetism 2e(London:Ox ford University Press)p150
[10]Stöh r J,Siegm ann H C(translated by Ji Y)2012 M agnetism:From Fundam en ta ls to Nanoscale Dynam ics (Beijing:H igher Education Press)p450(in Chinese) [Stöh r J,Siegm ann H C著 (姬扬 译)2012磁学:从基础知识到纳米尺度超快动力学(北京:高等教育出版社)第450页]
[11]Gabal M A,A ta-A llah S S 2004 J.Phys.Chem.Solids 65 995
[12]Li Y H,Kouh T,Shim IB,K im C S 2012 J.Appl.Phys. 111 07B 544
[13]Fayek M K,Sayed Ahm ed F M,A ta-A llah S S,E lnim er M K,M ostafa M F 1992 J.M ater.Sci.27 4813
[14]Lee D H,K im H S,Yo C H,Ahn K,K im K H 1998 M ater.Chem.Phys.57 169
[15]Saku rai S,Sasaki S,Okube M,Ohara H,Toyoda T 2008 Physica B 403 3589
[16]Harrison F W,Osm ond W P,Teale R W 1957 Phys. Rev.106 865
[17]Roum aih K 2011 J.M o l.Struct.1004 1
[18]Pervaiz E,Gu l IH 2012 J.Magn.M agn.M ater.324 3695
[19]Singhal S,Chand ra K 2007 J.Solid State Chem.180 296
[20]K adam R H,Birajdar A P,A lone S T,Shirsath SE 2013 J.M agn.M agn.M ater.327 167
[21]Srivastava M,Layek S,Singh J,Das A K,Verm a H C, O jha A K,K im N H Lee JH 2014 J.A lloys Com pd.591 174
[22]W ahba A M,M oham ed M B 2014 Ceram.Int.40 6127
[23]Iqbal M J,Ahm ad Z,M eydan T,M elikhov Y 2012 J. Appl.Phys.111 033906
[24]M ore S S,Kadam R H,Kadam A B,Shite A R,M ane D R,Jadhav K M 2010 J.A lloys Com pd.502 477
[25]Ghatage A K,Patil S A,Paran jpe S K 1996 Solid State Comm un.98 885
[26]Ida S,Ono K,Kozaki H(translated by Zhang Z X)1979 Data on Physics in Comm on Use (Beijing:Science Press)p133(in Chinese)[饭田修一,大野和郎,神前熙合编(张质贤译)1979物理学常用数表(北京:科学出版社)第133页]
[27]Tang G D,Han Q J,Xu J,Ji D H,Q i W H,Li Z Z, Shang Z F,Zhang X Y 2014 Physica B 438 91
[28]Xu J,Ji D H,Li Z Z,QiW H,Tang G D,Zhang X Y, Shang Z F,Lang L L 2015 Phys.Status Solidi B 252 411
[29]Xu J,M a L,Li Z Z,Lang L L,Q iW H,Tang G D,W u L Q,Xue L C,W u G H 2015 Phys.Status Solidi B 252 2820
[30]Xue L C,Lang L L,Xu J,Li Z Z,QiW H,Tang G D, W u L Q 2015 A IP Adv.5 097167
[31]D ing L L,Xue L C,Li Z Z,Li SQ,Tang G D,Q iW H, W u L Q,Ge X S 2016 A IP Adv.6 105012
[32]Shang Z F,QiW H,Ji D H,Xu J,Tang G D,Zhang X Y,Li Z Z,Lang L L 2014 Chin.Phys.B 23 107503
[33]Xu J,QiW H,Ji D H,Li Z Z,Tang G D,Zhang X Y, Shang Z F,Lang L L 2015 Acta Phys.Sin.64 017501 (in Chinese)[徐静,齐伟华,纪登辉,李壮志,唐贵德,张晓云,尚志丰,郎莉莉2015物理学报64 017501]
[34]Tang G D,Shang Z F,Zhang X Y,Xu J,Li Z Z,Zhen C M,Q iW H,Lang L L 2015 Physica B 463 26
[35]W u L Q,Q iW H,Li Y C,Li S Q,Li Z Z,Tang G D, Xue L C,Ge X S,D ing L L 2016 Acta Phys.Sin.65 027501(in Chinese)[武力乾,齐伟华,李雨辰,李世强,李壮志,唐贵德,薛立超,葛兴烁,丁丽莉2016物理学报65 027501]
[36]Phillips J C 1970 Rev.M od.Phys.42 317
[37]Ji D H,Tang G D,Li Z Z,Hou X,Han Q J,QiW H, Bian R R,Liu S R 2013 J.M agn.M agn.M ater.326 197
[38]Cohen R E 1992 Nature 358 136
[39]Cohen R E,K rakauer H 1990 Phys.Rev.B 42 6416
[40]W u L Q,Li Y C,Li S Q,Li Z Z,Tang G D,Q iW H, Xue L C,Ge X S,Ding L L 2015 AIP Adv.5 097210
[41]W u L Q,Li S Q,Li Y C,Li Z Z,Tang G D,Q iW H, Xue L C,D ing L L,Ge X S 2016 App l.Phys.Lett.108 021905
[42]Dupin J C,Gonbeau D,V inatier P,Levasseu r A 2000 Phys.Chem.Chem.Phys.2 1319
[43]Chen CW 1977M agnetism and M eta llurgy of Soft M agnetic M aterials(Am sterdam:North-Holland Pub lishing Com pany)p15
[44]Q iW H,M a L,Li Z Z,Tang G D,W u G H 2016 Acta Phys.Sin.66 027101(in Chinese)[齐伟华,马丽,李壮志,唐贵德,吴光恒2016物理学报66 027101]
[45]Shannon R D 1976 Acta Cryst.A 32 751
[46]Gou Q Q 1978 Introduction to Solid State Physics(Beijing:Peop le’s Education Press)p21(in Chinese)[苟清泉1978固体物理学简明教程(北京:人民教育出版社)第21页]
PACS:75.10.–b,75.47.Lx,75.47.NpDOI:10.7498/aps.66.067501
M olecu lar field origin for m agnetic ordering o fm agnetic m aterials∗
QiWei-Hua1)Li Zhuang-Zhi1)Ma Li1)2)Tang Gui-De1)2)†Wu Guang-Heng2)Hu Feng-Xia2)
1)(Hebei Advanced Thin Film Laboratory,College of Physics and Inform ation Engineering,Hebei Norm al University, Shijiazhuang 050024,China)
2)(State K ey Laboratory ofM agnetism,Institu te of Physics,Chinese Academ y of Sciences,Beijing 100190,China)
(Received 15 Novem ber 2016;revised m anuscrip t received 8 Decem ber 2016)
In 1907,Weiss proposed that there is a molecular field to exp lain the magnetic ordering of magnetic materials. However,it has not been clarified where them olecu lar field comes from so far.In recent decades,them agnetic ordering ofm etals and alloyswere exp lained by using the direct exchange interaction of between electrons on neighboring atom s, whilemagnetic ordering ofoxideswereexp lained by using the super exchange interaction and doubleexchange interaction m odels.The intrinsic relation between those exchange interactions has not been well exp lained.This resu lted in the fact that there arem any puzzles form agnetic ordering of them agneticm aterials.For exam p le,what role the Cr cations p lay in spinel ferrite CrFe2O4;why the calculated molecularmagneticmoment(3.85µB)for La0.85Sr0.15MnO3by using double exchange interaction m odel is lower than its experim ental value(4.20µB);whether there is a relation between the average atom m agnetic m om ent and their electrical resistivity for each of Fe,Co and Nim etals.These several puzzles have been exp lained recently by our group through using an O 2p itinerant electron model for magnetic oxides and a new itinerant electron m odel for m agnetic m etals.In this paper,a m odel for the m olecular field origin is p roposed. There are three states for the electrons rotating w ith high speed at the outer orbits of two ad jacent ions of m agnetic oxides or metals and alloys.1)There is a probability w ith which form the electron pairs w ith opposite spin directions and a certain life tim e,nam ed Weiss electron pairs(W EP);the static m agnetic attraction energy between two electrons of W EP is the elem entary origin of W eissm olecular field.2)There is a p robability w ith which two electrons w ith the same spin direction exchangemutually.3)If there are two electrons at the outer orbit of an ion,then for its ad jacent ion whose orbit has only one electron,the excess electron w ill itinerates between the ions.Furtherm ore,the energy equation of W EP,equilibrium distance,re0,and m aximum distance,rem,between electrons of W EP are derived.The probability w ith which WEP form s in each of several perovskitemanganites is investigated.For perovskitemanganites La0.8Ca0.2M nO3,La0.75Ca0.25M nO3,La0.70Sr0.30M nO3,the crystal cell constants increase linearly w ith tem perature when the tem perature is much lower than the Curie tem perature,TC,while they show a rapid increase nonlinearly near TC.We then calculate the diff erence in Mn—O bond length at TCbetween the linear and the non linear variation,∆dobs.Obviously,when the distance between the two electrons of W EP,re,is larger than the rem,W EP and the m agnetic ordering energy both disappear.Assum ing∆dobs=rem−re0,the probabilities w ith which W EP appears in La0.8Ca0.2MnO3,La0.75Ca.25MnO3,La0.70Sr0.30MnO3,are calculated to be 0.07%,0.31%and 3.13%,respectively. These resu lts indicate that the W EP m odel for them agnetic ordering energy is qualitatively reasonable.
magnetic ordermodel,magnetic oxides,magneticmetals and alloys
10.7498/aps.66.067501
∗国家自然科学基金(批准号:11174069)、河北省自然科学基金(批准号:A 2015205111)、河北省应用基础研究计划重点基础研究项目(批准号:16961106D)和河北省教育厅青年基金(批准号:QN 2016015)资助的课题.
†通信作者.E-m ail:tanggd@hebtu.edu.cn
*Pro ject supported by the National Natu ral Science Foundation of China(G rant No.11174069),the Natu ral Science Foundation of Hebei Province,China(G rant No.A 2015205111),the Key Item Science Foundation of Hebei Province,China (G rant No.16961106D),and the Young scholar Science Foundation of the Education Departm ent of Hebei Province,China (G rant No.QN 2016015).
†Corresponding author.E-m ail:tanggd@hebtu.edu.cn
猜你喜欢
环球时报(2022-04-18)2022-04-18
百科探秘·海底世界(2021年8期)2021-08-03
陶瓷学报(2020年6期)2021-01-26
航天器工程(2019年3期)2019-07-31
中学生数理化·中考版(2018年11期)2019-01-31
教学考试(高考化学)(2018年5期)2018-12-06
宝藏(2018年1期)2018-01-31
小主人报(2016年9期)2016-12-01
中国测试(2016年3期)2016-10-17
上海航天(2014年1期)2014-12-31
