
Br2分子在360—610 nm的光解离动力学研究∗
2017-10-23 07:16秦朝朝黄燕彭玉峰
物理学报 2017年19期
秦朝朝 黄燕 彭玉峰
1)(河南师范大学物理与材料科学学院,新乡 453007)
2)(光电子技术及先进制造河南省工程实验室,新乡 453007)
3)(河南师范大学电子与电气学院,新乡 453007)
Br2分子在360—610 nm的光解离动力学研究∗
秦朝朝1)2)†黄燕1)2)彭玉峰3)
1)(河南师范大学物理与材料科学学院,新乡 453007)
2)(光电子技术及先进制造河南省工程实验室,新乡 453007)
3)(河南师范大学电子与电气学院,新乡 453007)
利用包含转动自由度在内的含时薛定谔方程研究了Br2分子在波长范围为360—610 nm的光解离动力学.通过计算得到了Br2分子在四个特征波长处的切片解离影像,并经过分析得到了与切片解离影像相对应的动能分布;计算了Br2分子在波长范围为360—610 nm内总的动能分布,以及从A,B和C三个电子态解离的碎片各自所对应的动能分布;计算了A,B和C三个电子态各自的解离概率以及碎片产物的分支比Γ(Br∗/(Br+Br∗))随波长的变化.
光解离,解离影像,分支比
1 引 言
分子光解离动力学是分子反应动力学的一个重要组成部分[1−4].目前已有很多技术应用在这一领域,比如软X射线光电子能谱技术[5,6]、速度成像技术[7,8]、碎片离子成像技术[9,10]等.由于含溴化合物具有很强的破坏臭氧层的能力、及在灭火中具有重要作用[11,12]等,人们针对卤素分子和含有卤族元素的分子(如氯溴烷烃[13]、溴气[14]、四氟乙烷 [15]、氟利昂 [16−18]和氯碘烷烃[19]等)进行了广泛的研究.在360—610 nm内的连续吸收光谱中,可以将Br2光解离过程中产生的原子碎片分为两种:一种是基态的溴原子(用Br来表示),一种是自旋与轨道相耦合的激发态的溴原子(用Br∗来表示).Br2光解离动力学过程主要与四个电子态相关[20−22],其中包含一个基态和三个激发态这四个电子态可以通过四分量相对论的完全开壳层组态相互作用(COSCI)以及包含所有的单激发和双激发的多参考组态相互作用(MRCISD)方法计算得到[23].在这三个激发态的跃迁过程中,跃迁平行于分子轴[24],而这两个跃迁过程垂直于分子轴[25].科学家已采用共振增强多光子电离(REMPI)技术与时间切片的离子速度成像技术相结合的方法[26−28]研究了Br2的光解离过程.但是通过理论计算来分子解离影像进而分析光解离过程的报道还比较少.本文利用包含转动自由在内的含时薛定谔方程研究了Br2分子在波长范围为360—610 nm的光解离动力学.通过理论计算不仅可以给出总的动能分布,还可以给出A,B和C三个电子态所对应的动能分布.在Br2光解离的过程当中,利用动能随波长变化的分布规律,能够获得不同的解离通道达到最大值时所对应的峰值波长.在此基础上进一步计算了激发跃迁过程中解离碎片的分支比.运用分支比可以区分出在同一个跃迁过程中不同的光解离碎片通道.通过对Br2在360—610 nm波段内光解离过程所进行的一系列严格的定量分析和计算,可为其他类似分子光解离过程的探究提供依据.
2 理论方法
在计算Br2分子光解离动力学过程的理论模型中,主要与四个电子态相关,它们分别是Br2分子的电子基态和三个电子激发态如图1所示.
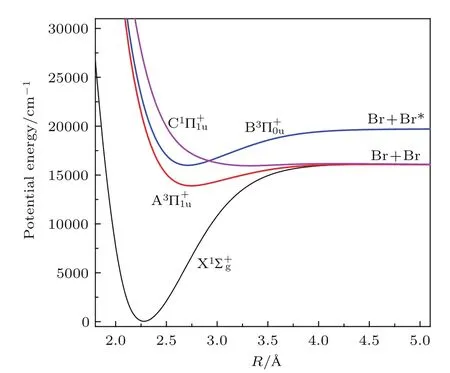
图1 (网刊彩色)Br2分子势能曲线图Fig.1.(color online)The relevant potential energy curves the Br2molecule.
为了方便起见,将这四个电子态简写为X,A,B和C.在波恩-奥本海默近似下可以通过求解包括转动自由度在内的含时薛定谔方程

来获得Br2分子的光解离动力学过程.其中ΨX(t),ΨA(t),ΨB(t)和ΨC(t)分别代表X,A,B和C四个电子态的波函数,R代表核间距,θ代表偏振方向与分子轴的夹角,ϕ代表方位角.在计算过程中,假定Br2分子最初的磁量子数M为零.由于线性极化场中磁量子数M是保守的,所以∂ϕ这一项可以忽略(其中ϕ是三维分子场哈密顿量中的方位角).此时,含时的三维分子场哈密顿量可以表示为[30]


其中µXA,µXB和µXC是跃迁偶极矩;m是约化质量;VX,VA,VB和VC分别代表X,A,B,C四个电子态在没有外场时的势能曲线.电场强度E(t)表达式为

其中E0,f(t),和ω分别代表电场强度最大值、载波包络形状和激光脉冲的中心频率.激光脉冲的载波包络形状f(t)是高斯函数f(t)=exp[−2ln2(t/τ)2],其中τ表示激光脉冲的半高宽度(full width at half minimum,FWHM).
通过计算动量空间中的输出粒子流[31,32],Br2分子与激光作用t时间后与波矢k,反冲方向有关的解离碎片分布可以表示为

其中Ra是渐进点,Rmax和Rmin为格点范围的最大值和最小值,波矢kn=2nπ/(Rmax−Rmin).需要指出的是,只有沿着解离方向(正方向)的动量才会对粒子流有贡献,而负方向部分可以忽略不计.在t时刻之前所有解离碎片的动量分布之和为

对所有解离方向的碎片求和可以得到解离碎片能量分布为

3 结果与讨论
计算中半径R由大小范围为[1.8,9.5]Å的1024个等距离网格来确定.为了排除其他非光解离过程反射进入内部区域的影响,在R>9.0 Å增加了吸收势.1024个等间距网格空间足够准确地描述Br2分子的光解离过程.60个高斯-勒让德正交点作为网络硬盘录像机的网格点(cosθ)数值来描述角动量自由度.假定包含转动自由度的各向同性的基态初始波函数为|v,J,M〉=|0,0,0〉.基态振动波函数是通过傅里叶哈密顿[33,34]得到的,然后采用劈裂算符方案[35,36]对波函数进行含时演化.当波包演化到达渐进区时,应有效地进行衰减,防止波包在边界反弹,造成干扰.采用吸收势Gabs(R)={1+exp[b∗(R−Rabs)]}−1来避免边界处的非物理反射,其中b=3 Å−1,Rabs=9 Å.
在计算过程中使用脉冲宽度FWHM为τ=30 fs,强度为I=1.0×1011W/cm2的线性偏振激光脉冲来解离Br2分子.
图2左侧给出了Br2分子在360—610 nm波长范围内四个特定波长所对应的光解离切片影像,可见圆环的半径随着波长的变化而变化.在激光波长为380 nm处的光解离影像中只有一个圆环,并且解离碎片主要分布在垂直于光的偏振方向上.这是因为激光波长为380 nm时只有Br2+hν→Br+Br这一个解离通道存在.在波长为460 nm和500 nm时光解离影像出现了第二个圆环,该圆环代表着另外一种光解离碎片的动量分布,该动量分布和与偏振方向平行的Br2+hν→Br+Br∗这个解离通道有关.然而在波长为560 nm时,从光解离影像又只观察到一个圆环,并且解离碎片与偏振方向垂直.从动能角度分析可知,这一圆环还是与Br2+hν→Br+Br这一解离通道相关.在每一个光解离切片影像对应的右侧是相应的总的动能分布,总的动能分布是通过对影像中所有角度进行积分得到的.针对以上的过程可以简单概括为:在波长为460 nm和500 nm的光解离影像中,半径稍小一点的内侧的圆环的与Br2+hν→Br+Br∗这一解离通道对应,而半径稍大一点的外侧圆环以及波长为380 nm与560 nm时的单个圆环的峰值都对应Br2+hν→Br+Br这一解离通道.
为了更详尽地描述Br2光解离过程,我们计算了360—610 nm波段内(间隔为1 nm)Br2分子光解离碎片的动能分布,如图3所示.容易发现,不仅总的解离概率与激光波长有关,Br+Br和Br+Br∗这两个不同的解离通道的碎片动能分布也依赖于激光波长.此外,解离碎片的动能随着波长的增大而减小.为了区分出不同电子态对总的光解离所产生的影响,计算了Br2三个激发态A,B,C所对应的动能分布,如图4所示.经过分析发现C电子态对Br2总的光解离概率贡献最大.而且不难发现,图3中总的动能分布和图4(c)中从C电子态解离得到的动能分布都在380 nm处达到最大值;图4(a)中从A电子态解离得到的动能分布在500 nm左右达到最大值;图4(b)中从B电子态解离得到的动能分布在460 nm左右达到最大值.动能分布的组分大小直观地反映了解离通道来源的比例.通过以上理论计算可以发现以下深层次解离信息:采用380 nm紫外光对Br2进行激发,该分子从C电子态发生解离的比例最大;采用500 nm可见光对Br2进行光激发,从A电子态发生解离的比例最大;而当采用460 nm可见光对Br2进行激发,发现该分子从B电子态发生解离的比例最大.

图2 (网刊彩色)激光波长为 (a)380 nm,(b)460 nm,(c)500 nm,(d)560 nm时的光解离碎片影像和相应的动能分布Fig.2. (color online)The slice imaging of dissociated fragments and the corresponding energydependent distribution P(E)for(a)380 nm,(b)460 nm,(c)500 nm,and(d)560 nm.

图3 (网刊彩色)光解离碎片的动能分布随着激光波长的变化Fig.3.(color online)The energy-dependent distribution P(E)of photofragments as a function of laser wavelength

图4 (网刊彩色)Br2三个激发态(a)对应的光解离碎片动能分布Fig.4.(color online)The energy-dependent distribution P(E)of Br fragments from excited state of
图5(a)给出了在激光波长360—610 nm范围内A,B和C三个电子态解离概率随波长的变化.对于第i个解离通道,解离概率的计算通过对相应的动能积分得到:

很明显,在波长λ<480 nm时A电子态的解离概率很小,且A电子态解离概率峰值只是C电子态解离概率峰值的13.0%,B电子态解离概率峰值是C电子态的43.4%.在长波范围内,它们的相对强度变得更加显著.通过分析发现,A电子态解离概率在波长为388 nm处达到最大值;B电子态在波长469 nm处其解离概率达到最大值;C电子态在波长为510 nm处其解离概率达到最大值.

图5 (网刊彩色)(a)三个激发态的解离概率;(b)分支比Γ(Br*/(Br+Br*))随波长的变化关系Fig.5.(color online)(a)The photodissociation probabilities for three excited states and(b)the branching ratio Γ(Br*/(Br+Br*))as a function of laser wavelength.
通过上述的讨论可以了解到不仅仅是总的解离概率与激光波长有关,每个电子态的解离概率也与激光波长有关.由于A和C两个电子态与Br+Br解离通道有关,而电子态B只与Br+Br∗解离通道有关,因此对于两个解离通道混合在一起的情况,可以通过分析解离通道的分支比来进一步研究.产生Br2+hν→Br+Br∗这一解离通道的分支比的定义式为
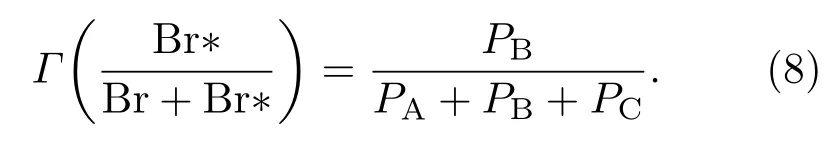
图5(b)给出了Br2+hν→Br+Br∗这一解离通道的分支比随着激光波长的变化曲线,并且发现在波长为462 nm时分支比达到最大值.另外,如图5(b)中所示,在波长较短λ<400 nm时分支比Γ(Br*/(Br+Br*))变得很小,几乎可忽略.这表明从C电子态光解离产生的Br+Br碎片远远多于从B电子态解离产生的Br+Br∗.然而在波长范围为λ=440—500 nm时光解离碎片Br+Br∗却又大大超过了Br+Br,因而B电子态的解离占据主导地位.在较长波段(λ>530 nm)时分支比Γ(Br*/(Br+Br*))同样也很低,接近于零,这表明这个波长范围内从A电子态产生的光解离碎片Br+Br占主导地位.如果在分支比的峰值忽略掉C电子态光解离的影响,分支比的峰值应为469 nm,这与考虑C电子态的影响时分支比所对应的峰值波长出现蓝移变为462 nm是符合的.由此可知,Br2的光化学反应通道与激发光的波长紧密相关,通过理论计算可以精确预测各个波长的光对解离通道的影响.该理论计算工作可以为通过调谐波长来精确控制Br2光化学反应通道提供重要的理论依据.
4 结 论
基于含时量子波包计算方法研究了Br2在近紫外360—610 nm内连续波段内的光解离.在Br2光解离过程中选取了四个有代表性的二维解离切片影像样本,影像中不同的圆环对应着不同的解离通道,解离影像圆环的半径与解离碎片的动能成正相关.同时也计算了在波长360—610 nm总的动能分布以及A,B和C三个电子态各自的动能分布.在此基础上找到A,B,C三个电子态最大解离概率所对应的峰值波长分别为510,469,388 nm.A电子态的解离概率的峰值强度相对于C电子态为13.0%,而B电子态相对于C电子态为43.4%.除此之外,计算得到了在所研究波段的解离碎片的分支比,发现在波长为462 nm时分支比达到最大值.本文对近紫外波段范围内Br2光解离的计算和研究得到了许多有意义的信息,从计算上进一步分析了其过程机理并给出了合理的解释.研究表明,可以通过分支比来确定不同电子态解离碎片或解离通道的组成以及参照能量分布随波长的变化分布来确定光解离过程中不同电子态的解离概率.
[1]Davies J A,LeClaire J E,Continetti R E,Hayden C C 1999J.Chem.Phys.111 1
[2]Ashfold M N R,Baggott J E 1989Molecular Photodissociation Dynamics(Letchorth:Wiley Press)p243
[3]Demyanenko A V,Potter A B,Dribinski V,Reisler H 2002J.Chem.Phys.117 2568
[4]Rakitzis T P,Kitsopoulos T N 2002J.Chem.Phys116 9228
[5]Nugent-Glandorf L,Scheer M,Samuels D A,Mulhisen A M,Grant E R,Yang X M,Bierbaum V M,Leone S R 2001Phys.Rev.Lett.87 1103
[6]Nugent-Glandorf L,Scheer M,Samuels D A,Bierbaum V M,Leone S R 2002J.Chem.Phys.117 1063
[7]Klemm A,Kimmich R,Weber M 2001Phys.Rev.E63 041514
[8]Han S I,Pierce K L,Pines A 2006Phys.Rev.E74 016302
[9]Rogers L J,Ashfold M N R,Matsumi Y,Kawasaki M Whitaker B J 1996Chem.Phys.Lett.258 159
[10]Beckert M,Greaves S J,Ashfold M N R 2003Phys.Chem.Chem.Phys.5 308
[11]Kato H,Baba M 1995Chem.Rev.95 2311
[12]Asano Y,Yabushita S 2003Chem.Phys.Lett.372 348
[13]Liu Y Z,Xiao S R,Zhang C Y,Zheng G G,Chen Y Y 2012Acta Phys.Sin.61 193301(in Chinese)[刘玉柱,肖韶荣,张成义,郑改革,陈云云2012物理学报61 193301]
[14]Zhang J,Zhang S A,Yang Y,Sun S Z,Wu H,Li J,Chen Y T,Jia T Q,Wang Z G,Kong F N,Sun Z R 2014Phys.Rev.A90 053428
[15]Kettunen J A,Sankari A,Partanen L,Urpelainen S,Kivimäki A,Huttula M 2012Phys.Rev.A85 062703
[16]Liu Y Z,Chen Y Y,Zheng G G,Jin F,Knopp G 2016Acta Phys.Sin.65 053302(in Chinese)[刘玉柱,陈云云,郑改革,金峰,Knopp Gregor 2016物理学报65 053302]
[17]Liu Y Z,Xiao S R,Wang J F,He Z F,Qiu X J,Knopp G 2016Acta Phys.Sin.65 113301(in Chinese)[刘玉柱,肖韶荣,王俊锋,何仲福,邱学军,Knopp Gregor 2016物理学报65 113301]
[18]Liu Y Z,Deng X L,Li S,Guan Y,Li J,Long J Y,Zhang B 2016Acta Phys.Sin.65 193301(in Chinese)[刘玉柱,邓绪兰,李帅,管跃,李静,龙金友,张冰2016物理学报65 193301]
[19]Liu Y Z,Long J Y,Xu L X,Zhang X Y,Zhang B 2017Chin.Phys.Lett.34 033301
[20]Orr-Ewing A J 2015Ann.Rev.Phys.Chem.66 119
[21]Orlando J J,Burkholder J B 1995J.Phys.Chem.99 1143
[22]Tellinghuisen J 2001J.Chem.Phys.115 10417
[23]Gomes J D,Gargano R,Martins J B L,de Macedo L G M 2014J.Phys.Chem.A118 5818
[24]Focsa C,Li H,Bernath P F 2000J.Mol.Spectrosc.200 104
[25]Yukiya T,Nishimiya N,Samejima Y,Yamaguchi K,Suzuki M,Boone C D,Ozier I,Le Roy R J 2013J.Mol.Spectrosc.283 32
[26]Jung Y J,Park M S,Kim Y S,Jung K H 1999J.Chem.Phys.111 4005
[27]Kim T K,Park M S,Lee K W,Jung K H 2001J.Chem.Phys.115 10745
[28]Zhu R S,Tang B F,Zhang X,Zhang B 2010J.Phys.Chem.A114 6188
[29]Han Y C,Yuan K J,Hu W H,Yan T M,Cong S L 2008J.Chem.Phys.128 134303
[30]Numico R,Keller A,Atabek O 1995Phys.Rev.A52 1298
[31]Jolicard G,Atabek O 1992Phys.Rev.A46 5845
[32]Jolicard G,Billing G D 1991Chem.Phys.149 261
[33]Marston C C,Balintkurti G G 1989J.Chem.Phys.91 3571
[34]Willner K,Dulieu O,Masnou-Seeuwsa F 2004J.Chem.Phys.120 548
[35]Bandrauk A D,Shen H 1993J.Chem.Phys.99 1185
[36]Chu T S,Zhang Y,Han K L 2010Int.Rev.Phys.Chem.25 201
Photodissociation dynamics of Br2in wavelength range of 360–610 nm∗
Qin Chao-Chao1)2)†Huang Yan1)2)Peng Yu-Feng3)
1)(College of Physics and Materials Science,Henan Normal University,Xinxiang 453007,China)
2)(Engineering Laboratory for Optoelectronic Technology and Advanced Manufacturing of Henan Province,College of Physics and Electronic Engineering,Henan Normal University,Xinxiang 453007,China)
3)(College of Electronic and Electrical Engineering,Henan Normal University,Xinxiang 453007,China)
We study the photodissociation of Br2in a wavelength range from 360 nm to 610 nm in the near-visible UV continuum band based on the calculation of time-dependent quantum wave packet including the rotational degree of freedom.We calculate four representative samples of two-dimensional(2D)slice images taken from photolysis of Br2molecules,in which the di ff erent rings in the 2D slice images are corresponding to the di ff erent photodissiation channels.The radius of each 2D slice image ring is positively related to kinetic energy of photofragment.The maximum photofragment flux perpendicular or parallel to the photolysis polarization is also related to photodissiation channel.Furthermore,we calculate the total kinetic energy distributionP(E)and theP(E)distribution from the respective electronic excited states A,B and C in the wavelength range of 360–610 nm,from which we fi nd that the wavelengths corresponding to the maximum dissociation probability from respective electronic excited states A,B and C are 510 nm,469 nm,and 388 nm,respectively.As is well known,not only the total dissociation probability,but also the respective dissociation probability of electronic excited states is dependent on the laser wavelength.We also calculate the dissociation probabilities from electronic excited states A,B and C,respectively.We fi nd that the dissociation probability of electronic excited state A is not signi fi cant whenλ<480 nm and that the peak intensity of the dissociation probability to the A state is about 13.0%of that to the C state,while that to the B state is about 43.4%.In addition,because the electronic excited states A and C are related to the photodissociation channel Br+Br,and the electronic excited state B is corresponding to the photodissociation channel Br+Br*,the images which reveal the involvement of more than one product channel can be analyzed by the respective channel branching ratios.At the short wavelength(λ<400 nm)the branching ratioΓ(Br*/(Br+Br*))is small,even near to zero,which re fl ects that electronic state C transition gives rise to many Br+Br over Br+Br*.However,within the wavelength range(λ=440–500 nm)Br+Br*photofragments are excess of Br+Br,so the electronic state B transition is dominant.At longer wavelength(λ>530 nm)the branching ratioΓ(Br*/(Br+Br*))is also low,near to zero,indicating the prevalence of electronic state A transition.Ignoring the dissociation from electronic state C,the maximum dissociation probability 469 nm is consistent with branching ratio maximum 462 nm.Because the electronic excited state C is related to the photodissociation channel Br+Br,the branching ratio will be reduced.So the maximum wavelength of branching ratio is blue shifted.
photodissociation,slice image,branching ratio
8 May 2017;revised manuscript
5 July 2017)
(2017年5月8日收到;2017年7月5日收到修改稿)
10.7498/aps.66.193301
∗国家自然科学基金(批准号:U1404112,11404411)、河南省科技攻关研究项目(批准号:142102310274,172102210340)和河南省教育厅重点项目(批准号:17A140021)资助的课题.
†通信作者.E-mail:qinch@hotmail.com
©2017中国物理学会Chinese Physical Society
PACS:33.80.Gj,33.20.Lg,82.80.Ms
10.7498/aps.66.193301
*Project supported by the National Natural Science Foundation of China(Grant Nos.U1404112,11404411),the Advanced Technology Research Program of Henan Province,China(Grant Nos.142102310274,172102210340),and the Foundation for Key Program of Education Department of Henan Province,China(Grant No.17A140021).
†Corresponding author.E-mail:qinch@hotmail.com
猜你喜欢
金桥(2022年4期)2022-05-05
黑龙江大学自然科学学报(2022年1期)2022-03-29
数学物理学报(2021年4期)2021-08-30
学生天地(2019年28期)2019-08-25
人大建设(2018年7期)2018-09-19
商周刊(2018年15期)2018-07-27
商周刊(2018年11期)2018-06-13
中南大学学报(自然科学版)(2016年2期)2017-01-19
中国照明(2016年4期)2016-05-17
中国当代医药(2015年26期)2015-03-01
