
慈竹叶蝉类害虫DNA条形码分析
2018-03-26 05:51姚亚林罗强董梦书陈祥盛杨琳
四川动物 2018年2期
姚亚林, 罗强, 董梦书, 陈祥盛, 杨琳*
1. 贵州大学林学院,贵阳550025; 2. 贵州大学昆虫资源开发利用省级特色重点实验室,贵阳550025; 3. 贵州大学昆虫研究所,贵阳550025; 4. 贵州大学贵州山地农业病虫害省级重点实验室,贵阳550025)
叶蝉是半翅目Hemiptera叶蝉科Cicadellidae物种的统称,其种类多、数量大,是农林害虫中的重要类群。西南地区是我国也是世界竹类生产和分布中心区域之一。近年调查发现,西南地区慈竹Bambusaemeiensis叶蝉类害虫种类多、数量大且分布广,危害严重。对该地区竹林叶蝉类害虫进行多样性研究是林业生态系统保护和林业资源管理的共同需求,但叶蝉类害虫的物种鉴定一直是多样性研究的难点,主要原因有:(1)叶蝉类昆虫体型和体色等鉴别特征的形态性状复杂多变,易随发育阶段或生境的改变而产生较大变化,如额垠叶蝉属Mukaria种类(Yang & Chen,2011),传统形态鉴定容易造成同种异名或异种同名等错误;(2)由于缺乏对竹类叶蝉整个生活史的了解,不同虫态叶蝉物种的鉴定较困难。近年来,随着分子生物学技术的发展,分子鉴定技术为物种分类与鉴定研究提供了有效手段。
DNA条形码技术的出现极大方便了物种鉴定工作(Hebertetal.,2003,2004a)。在大多数动物类群中,线粒体细胞色素C氧化酶第一亚基(COⅠ)基因被广泛采纳为通用的标准条形码基因片段(Hebertetal.,2004b;Wardetal.,2005;Hajibabaeietal.,2006;Ratnasinghametal.,2007)。然而在有些动物类群中,COⅠ基因并不是唯一适合的条形码片段,如Sinniger等(2008)对六放珊瑚目Zoanthidea及Stampar等(2012)对角海葵目Cerianthidea的研究发现,COⅠ和16SrRNA基因均可准确区分近缘种,实现物种的准确鉴定;亦有研究表明,一些类群(如角顶叶蝉类Deltocephalus-like、弯钩叶蝉属Flexamia、黄翅叶蝉属Dalbulus)的16SrRNA基因序列片段可用于区分近缘种(Fangetal.,1993;Dietrich,1997),可见不同动物类群的最适条形码并非一致。在叶蝉类昆虫的DNA条形码研究中,相比于COⅠ基因,16SrRNA基因序列片段较易扩增,常被作为一种有效的分子标记,用于区分形态近似种或描述新发现的种类(Schuchert,2006;Migliettaetal.,2009;Mirandaetal.,2010;Mouraetal.,2011a,2011b),然而,我国西南地区竹林叶蝉科昆虫DNA条形码的相关研究尚未有报道。
本文以中国常见竹种慈竹为寄主的叶蝉类昆虫为研究对象,扩增其COⅠ和16SrRNA基因序列片段,比较两者作为标准条形码基因片段的适用性和潜力,同时,结合构建系统发育树和矢量分析法评估这2个基因序列片段在竹子叶蝉分子鉴定中的应用前景,为竹林叶蝉类昆虫的准确快速鉴定提供一定的参考。
1 材料与方法
1.1 供试叶蝉
实验叶蝉于2014年5月—2016年11月采集于贵州、四川、云南、重庆4个省市的26个地区,传统形态分类方法鉴定,采集及物种鉴定信息如表1所示。采集的成虫浸泡于1.5 mL离心管中,第一个月更换无水乙醇3~4次(Sunetal.,2013),后放置于-70 ℃超低温冰箱中长期保存待用(Zhang & Kang,2001;Zhangetal.,2004),其余未经酒精浸泡的标本干燥处理后保存于贵州大学昆虫研究所标本馆。
1.2 试剂及仪器
昆虫DNA提取试剂盒购于Omega BioTek(D0926-01);引物由生物工程(上海)股份有限公司合成;Olympus SZ2-ILST型光学体视显微镜(德国Leica);T100TMThermal Cycler型PCR扩增仪(美国Bio-Rad);170-8170型UVP凝胶成像系统(美国Bio-Rad);Power PacTMHV Power Supply型电泳仪(美国Bio-Rad);Mini-10K微型高速离心机(珠海黑马医学仪器有限公司);HVE-50型自动高压灭菌器(日本HIRAYAMA)。
1.3 DNA提取及PCR扩增
标本解冻后,将腹部放入甘油中保存,剩余的虫体取头、胸、足等部分研磨至粉末状,然后用Omega BioTek生产的E.Z.N.A.TMInsect DNA kit试剂盒提取总DNA,产物-20 ℃保存。
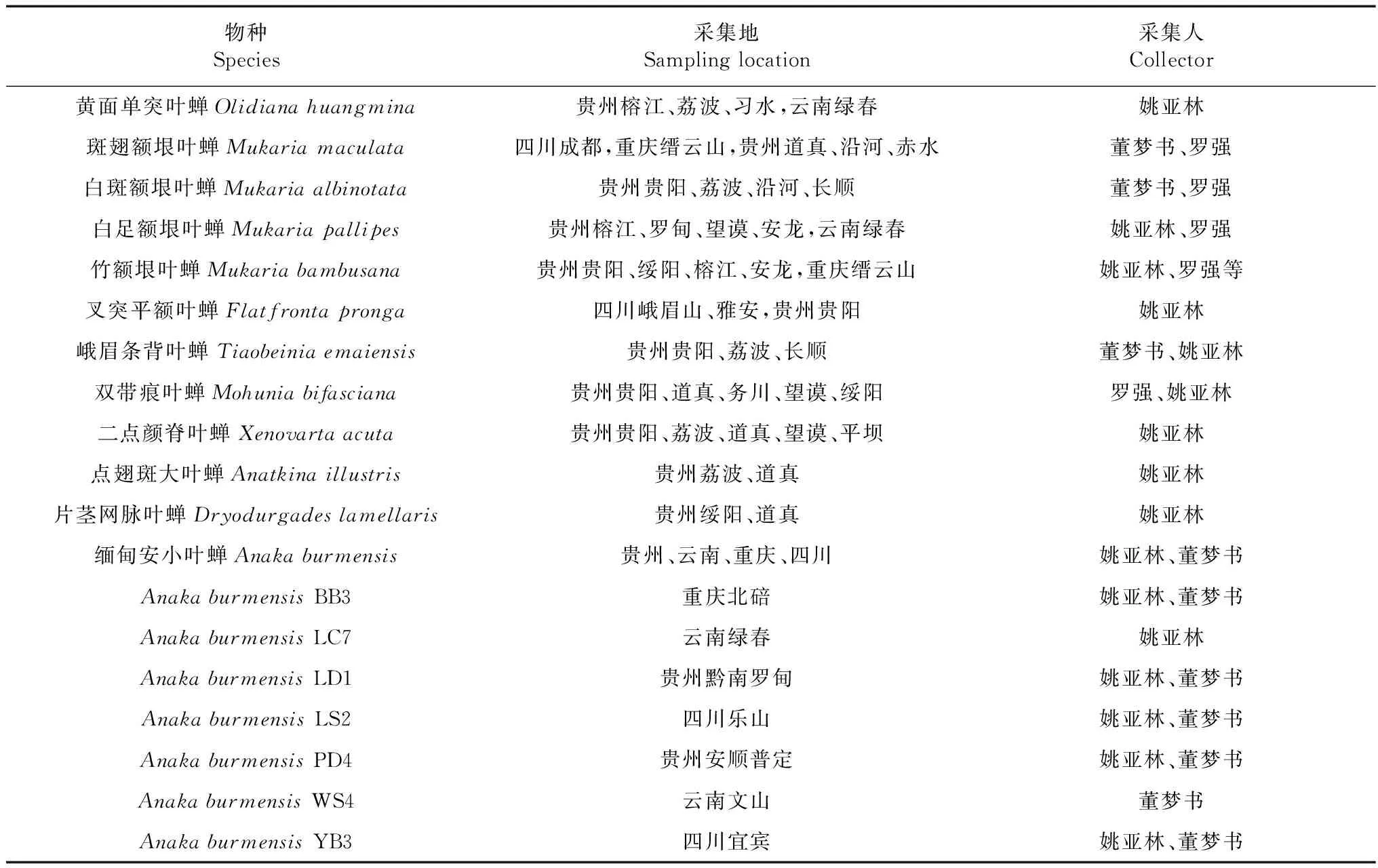
表1 物种采集信息Table 1 Information of sampling
注: BB3. 重庆北碚, LC7. 云南绿春, LD1. 贵州黔南罗甸, LS2. 四川乐山, PD4. 贵州安顺普定, WS4. 云南文山, YB3. 四川宜宾
Notes: BB3. Beibei district of Chongqing, LC7. Lvchun county, Yunnan province, LD1. Luodian county, Guizhou province, LS2. Leshan city, Sichuan province, PD4. Puding county, Guizhou province, WS4. Wenshan city, Yunnan province, YB3. Yibin city, Sichuan province
以提取的总DNA为模板进行序列的PCR扩增,反应程序参照Folmer等(1994),扩增引物分别为:16SrRNA-F:5’-CCGGTYTGAACTCARATCAWGT-3’,16SrRNA-R:5’-CTGTTTAWCAAAAACATTTC-3’;COⅠ-F:5’-GGTCAACAAATCATAAAGATATTG-3’,COⅠ-R:5’-TAAACTTCAGGGTGACCAAAAAAT-3’。PCR反应体系为30 μL:模板DNA 3 μL,上下游引物各1 μL,Taq PCR Master Mix 15 μL,ddH2O补至30 μL。PCR扩增程序:94 ℃预变性3 min;94 ℃变性30 s,50 ℃退火30 s,72 ℃延伸1 min,33个循环;最后72 ℃延伸10 min,4 ℃保存。PCR产物取3 μL采用1%琼脂糖凝胶电泳检测,纯化步骤参照DNA凝胶回收试剂盒Omega BioTek(D0926-01)程序。回收产物送生工生物工程(上海)股份有限公司双向测序。
1.4 数据分析
将获得的测序峰图利用DNAstar 5.0进行正反链校对和编辑,手动去除序列两端的引物区,获得有效片段的样本序列。Meglign进行排序后利用Clustal X进行多序列比对,采用DnaSP v5计算信息位点、变异位点数,通过MEGA 6.06分析碱基组成、变异位点,基于Kimura-2-parameter(K2P)模型进行遗传距离分析,采用邻接法(Neighbor-Joining,NJ)构建系统发育树,节点支持率采用自展值进行估计,重复检验1 000次。选取蝉总科蝉科Cicadoidea的日本寒蝉Meimunaopalifera(16SrRNA基因序列GenBank登录号为AY139968.1)及蜡蝉总科Fulgoroidea飞虱科Delphacidae的四刺小头飞虱Malaxellatetracantha(COⅠ基因和16SrRNA基因序列GenBank登录号分别为HM233883.1和HM233744.1)和黄小头飞虱Malaxellaflava(COⅠ基因和16SrRNA基因序列GenBank登录号分别为HM233896.1和HM233772.1)共6条序列作为外群。参照Sirovich等(2009,2010)的矢量分析方法,构建中国慈竹常见叶蝉科昆虫条形码Klee-diagram矢量图。
2 结果与分析
2.1 序列分析
经通用引物PCR扩增、序列测定,共获得COⅠ和16SrRNA基因序列45条,其中,COⅠ基因序列4科 5属8种21条,GenBank登录号为MG376677~MG376697;16SrRNA基因序列3科7属9种24条,GenBank登录号为MG376698~MG376721。利用MEGA 6.06对21条竹类叶蝉COⅠ基因序列进行比对后保留了590个的同源性序列,分析表明,在590个位点中没有插入和缺失,总体序列的T、C、A、G平均含量分别为36.20%、12.70%、41.10%、10.00%,A+T含量较高,为77.30%;保守位点、变异位点和简约信息位点分别为310个、280个和237个,在590个位点中分别占52.54%、47.46%和40.17%。对24条16SrRNA基因序列进行比对后保留了463 bp的同源性序列,分析表明,在463个位点中存在59个插入/缺失位点,总体序列的T、C、A、G平均含量分别为35.60%、15.10%、41.20%、8.10%,A+T含量较高,为76.80%;保守位点、变异位点和简约信息位点分别为235个、221个和192个,在463个位点中分别占50.76%、47.73%和41.47%。COⅠ基因和16SrRNA基因的T、C、A、G含量不同,但均呈现出明显的A+T偏向性,特别是COⅠ基因密码子第三位C、G平均含量在不同物种间差异很大。
2.2 遗传差异
以K2P模型计算序列间遗传距离,并按照分类阶元进行统计分析(表2~表4)。结果显示,16SrRNA基因序列的种内遗传距离为0~0.010(均值为0.003),而同科不同种间的遗传距离为0.151~0.396(均值为0.257),其中,斑翅额垠叶蝉M.maculata和白斑额垠叶蝉Mukariaalbinotata间的遗传距离最小,为0.151,叉突平额叶蝉Flatfrontapronga和缅甸安小叶蝉Anakaburmensis间的遗传距离最大,为0.396。COⅠ基因的种内遗传距离为0~0.013(均值为0.004),其中84%的种内遗传距离小于0.003(图2),这可能与扩增的叶蝉样本中缅甸安小叶蝉和额垠叶蝉群体的序列所占比重大,而其他叶蝉只有单条序列有关;缅甸安小叶蝉云南绿春种群(LC7)与其他种群间出现种内遗传距离的最大值(0.013);COⅠ基因的种间遗传距离为0.159~0.341(均值为0.283),斑翅额垠叶蝉和白斑额垠叶蝉间的遗传距离最小(0.159);2个基因序列在所调查种类中,种内遗传差异均小于种间遗传差异,存在明显的条形码间隔(图1,图2)。
2.3 基于系统发育树的物种鉴定
基于COⅠ和16S rRNA基因序列的系统发育NJ树中,2个基因片段所有同种的序列都能形成具有较高支持度的单分支(图3,图4)。小叶蝉亚科Typhlocybinae、圆痕叶蝉亚科Megophthalminae、离脉叶蝉亚科Coelidiinae和角顶叶蝉亚科Deltocephalinae的分支证明COⅠ和16SrRNA基因序列均能很好地区分物种。在更高的分类阶元中,16SrRNA基因序列可将额垠叶蝉族Mukariini中的额垠叶蝉属(斑翅额垠叶蝉、白斑额垠叶蝉、白足额垠叶蝉M.pallipes、竹额垠叶蝉Mukariabambusana)成功分为单性孔组和双性孔组两大支,二点颜脊叶蝉Xenovartaacuta与(叉突平额叶蝉+峨眉条背叶蝉Tiaobeiniaemaiensis+双带痕叶蝉Mohuniabifasciana)这一支系也能较好分开(图3);而COⅠ基因序列在额垠叶蝉族的系统发育关系中(图4),额垠叶蝉属与叉突平额叶蝉、峨眉条背叶蝉、双带痕叶蝉也能很好分开,且也成功分为单性孔组和双性孔组两大支,证明COⅠ基因序列亦能有效区分物种。
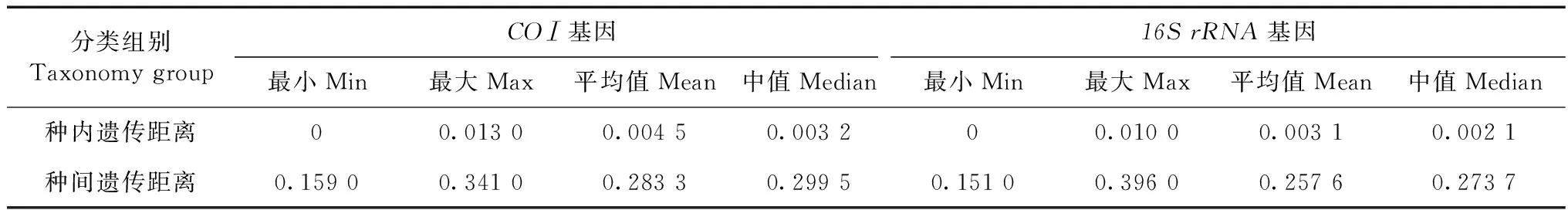
表2 叶蝉类各分类阶元遗传距离统计Table 2 Genetic distance on different taxonomic levels of leafhoppers

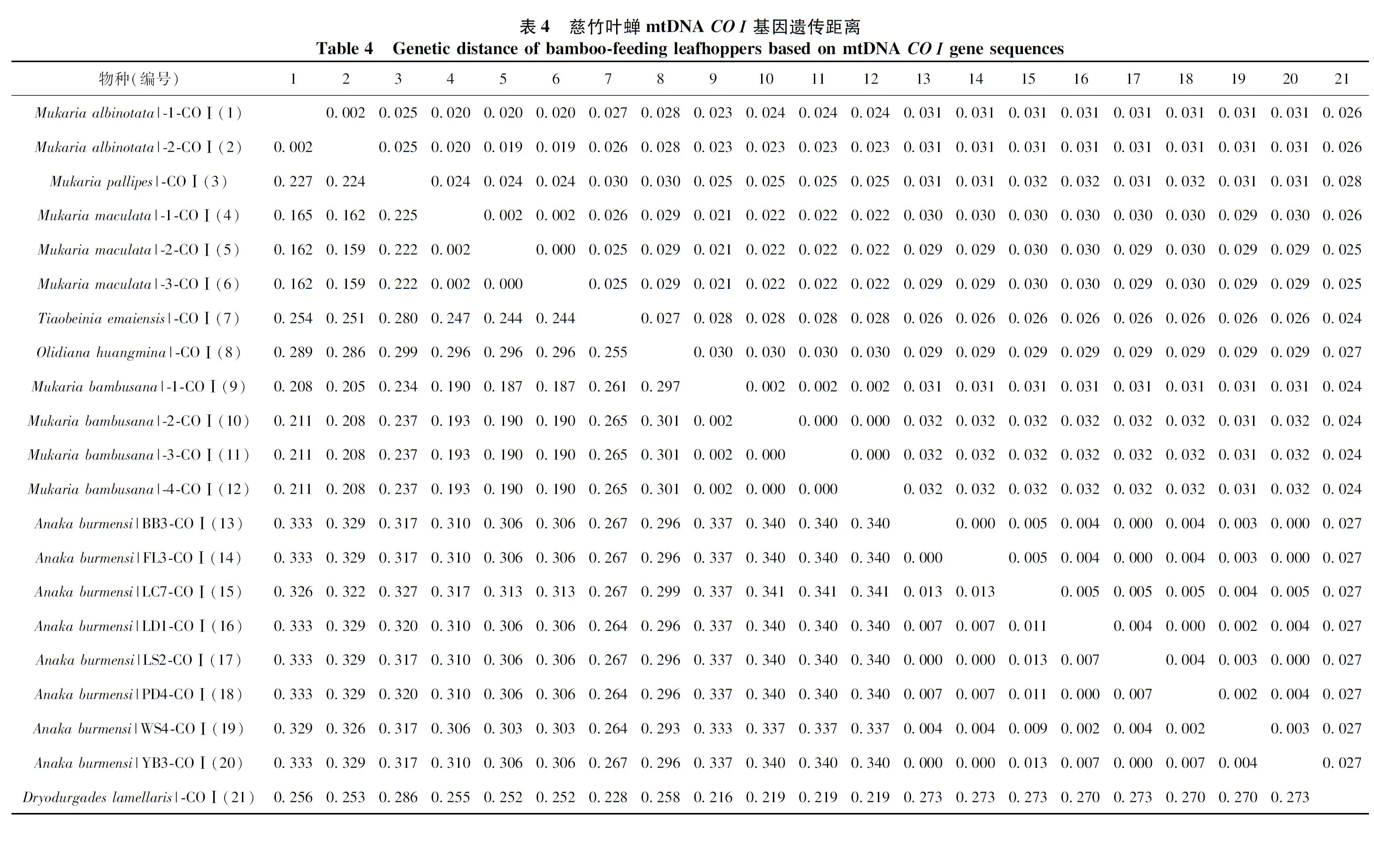

图1 16S rRNA基因序列片段的种内与种间K2P遗传距离的分布频率Fig. 1 Frequency distribution of the K2P intra- and inter-species genetic distances for 16S rRNA gene sequences
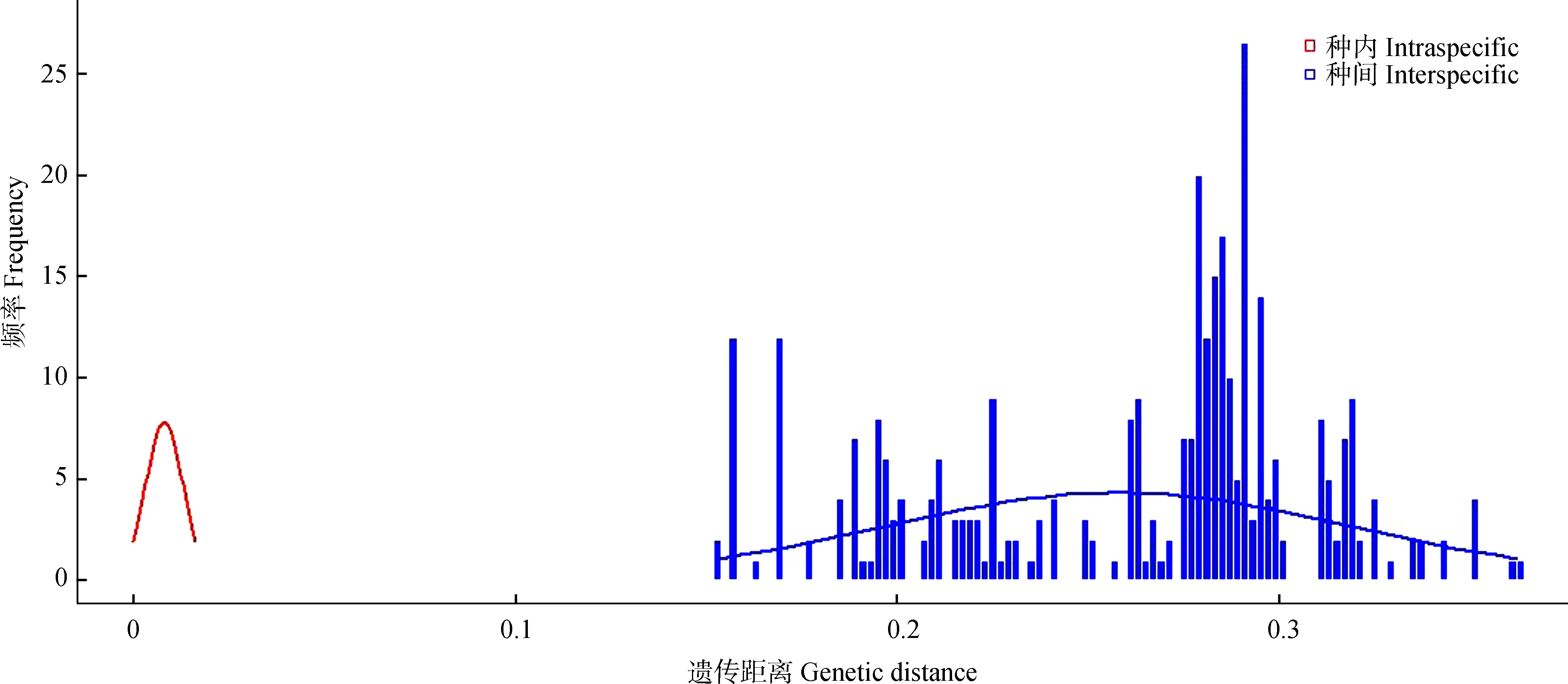
图2 COⅠ基因序列片段的种内与种间K2P遗传距离的分布频率Fig. 2 Frequency distribution of the K2P intra- and inter-species genetic distances for COⅠ gene sequences
2.4 基于Klee-diagram的矢量分析
基于西南地区常见慈竹叶蝉的矢量分析发现,COⅠ和16SrRNA基因片段均显示明显的种内序列比对“热区”,即种内序列相似程度明显高于种间序列,直观显示出种内、种间遗传距离差异;2个片段的缅甸安小叶蝉序列均表现为独立的“热区”,显示出其与角顶叶蝉亚科额垠叶蝉族、大叶蝉亚科Cicadellinae序列的明显差别,这与NJ树分析结果一致(图5,图6)。
3 讨论
理想的条形码种内遗传差异会小于近缘物种种间差异(Del-Pradoetal.,2010),即种内、种间遗传差异的幅度确定了物种的界限(Köhler,2007)。若种内、种间遗传差异存在重叠,则说明该分子标记无法准确区分物种。理论上,有效的DNA条形码应存在条形码间隔,即种内、种间遗传距离差异明显。
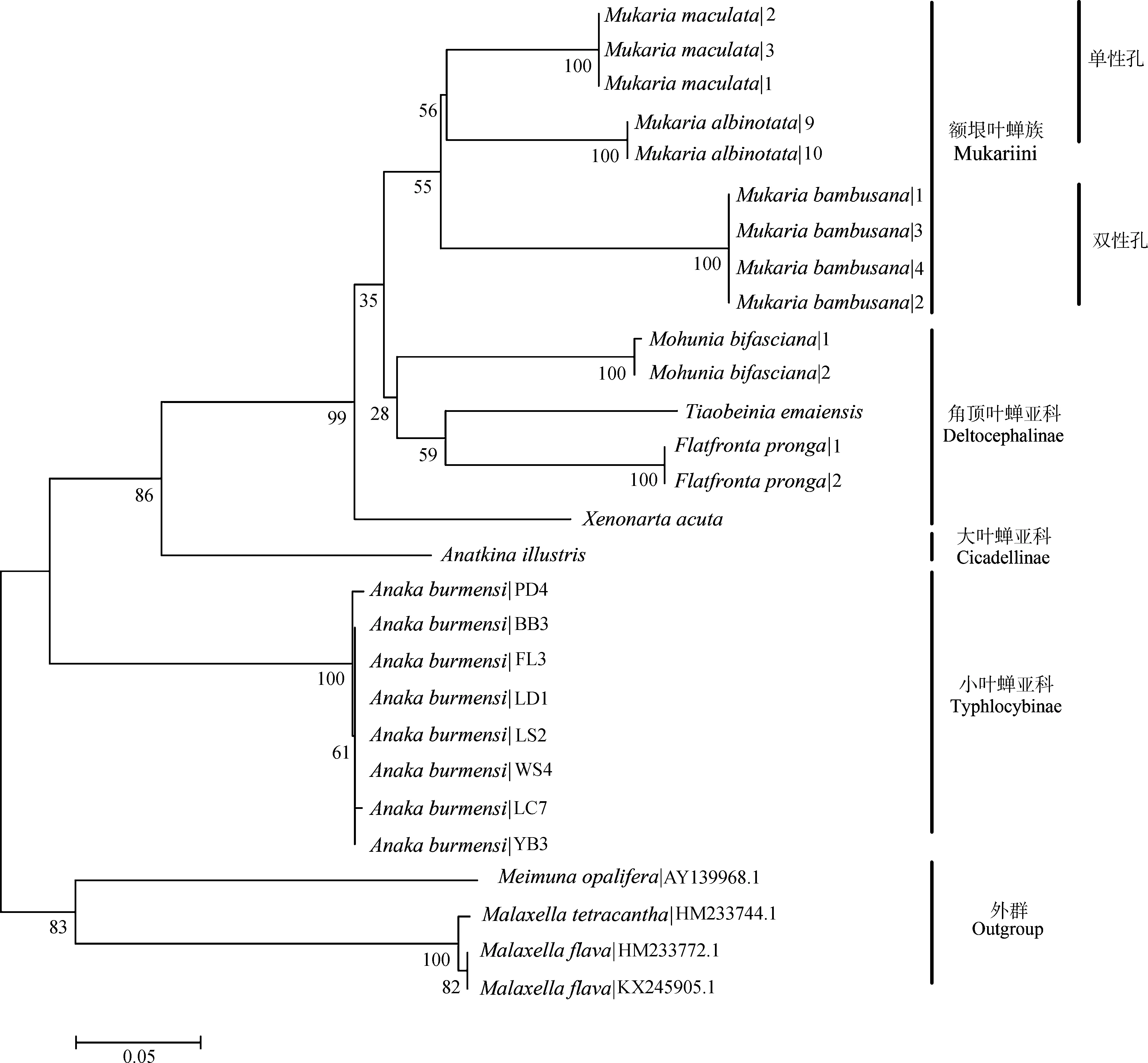
图3 基于16S rRNA基因序列的邻接树Fig. 3 Neighbor-Joining clustering based on 16S rRNA gene sequences
Hebert等(2003)提出,种间遗传差异应达到种内遗传差异的10倍。有研究证实,水螅纲Hydrozoa多个类群的COⅠ基因序列片段种内、种间遗传距离并不重叠(Zemlaketal.,2009;Ortmanetal.,2010;Sunetal.,2012)。本研究所涉及种类的COⅠ和16SrRNA基因序列片段均有明显条形码间隔,未出现种内、种间遗传差异存在重叠的种群。2个片段同属种间的遗传距离均比种内高出30倍,证实了它们作为慈竹叶蝉类昆虫DNA条形码标准基因的可行性。结果出现高30倍的主要原因可能是本研究是对寄主慈竹上有分布的叶蝉科昆虫标本进行采样扩增,而叶蝉科昆虫还有其他寄主,因受寄主范围的限制致使出现这一结果。
本研究针对西南地区慈竹常见叶蝉类昆虫,基于K2P遗传距离和NJ树的方法证明了COⅠ和16SrRNA基因序列片段均可有效运用于DNA条形码研究,这一结果可促进叶蝉类的生物多样性研究。但基于NJ树鉴定物种时,应考虑该方法是基于距离类的建树方法,在序列分歧较大的数据中估计的序列间遗传距离方差较大,这将对系统发育树拓扑分
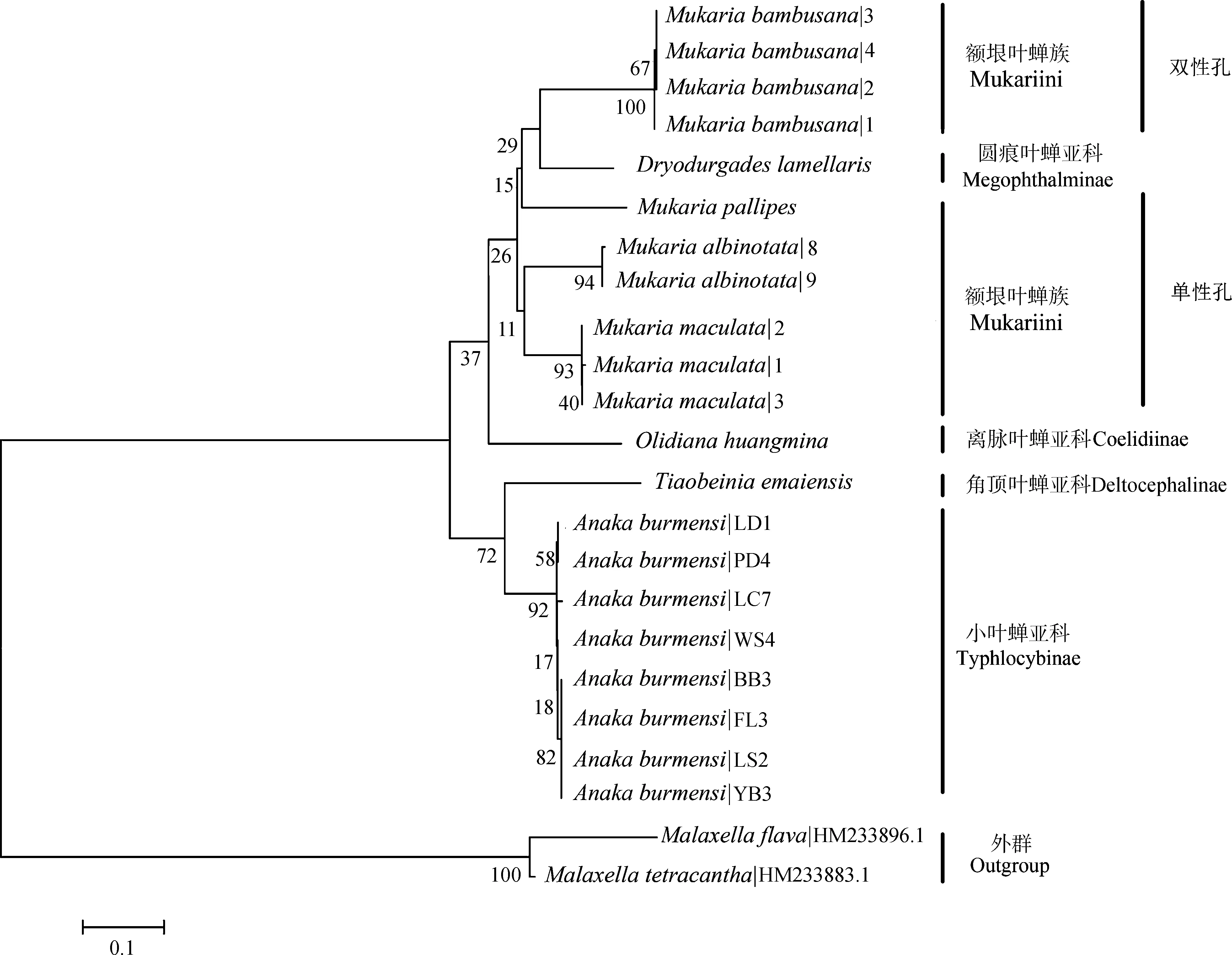
图4 基于COⅠ基因序列的邻接树Fig. 4 Neighbor-Joining clustering based on COⅠ gene sequences
支结构和支长估计带来影响(Yang & Rannala,2010),而本文研究对象类群跨度也较大,可能存在类似的影响。自Sirovich等(2009,2010)引入矢量分析后,Klee-diagram有效地促进了DNA条形码的应用,但Klee-diagram矢量分析只将未知序列划归到确定的种、属等动物类群,并未对序列的演化状态做出推断(Sirovichetal.,2009;Stoeckle & Coffran,2013),因此,不能从系统发育与演化的角度来评价和讨论Klee-diagram矢量分析的鉴定效率。依本研究中Klee-diagram矢量分析结果,16SrRNA基因序列的鉴定效率较优于COⅠ基因,因样本量限制等原因,这种结果可能不具有普遍性,下一步还需竹类叶蝉科昆虫大样本数据验证。尽管该方法并未给出明晰的谱系关系,但因适宜大样本数据和可视化界面等,其在农林昆虫的准确鉴定中仍有较大的应用潜力。
尽管如此,基于种内种间遗传差异、矢量分析的类群划分和系统发育树分析等方法在农林叶蝉类昆虫的多样性研究中还将发挥更大的作用。由于本研究获取的基因片段数量和寄主范围局限,未能在更广泛的地理范围内讨论竹类叶蝉科昆虫的遗传差异和隐种分化对这2个片段鉴定效率的影响。今后随着调查和采集的深入,竹类叶蝉科昆虫标本的增加以及其他候选条形码基因片段的加入,可能呈现出不同遗传差异,出现种内、种间遗传差异存在重叠的种群,因此,使用种内种间遗传差异达到10倍阈值鉴定不同物种的方法受到限制;若利用不同候选条形码基因片段构建系统发育树鉴定物种,有可能出现基因树冲突问题,因此,应当考虑多种方法整合分析和评价不同候选条形码基因片段的鉴定功效。
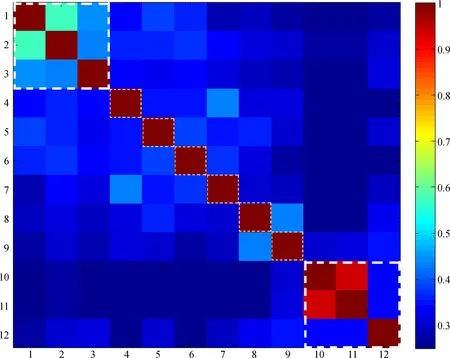
图5 16SrRNA基因条形码矢量图
Fig. 5 Klee-diagram for the 16SrRNAgene
横纵坐标均为序列编号; 彩色图例自冷色至暖色表示序列相似性增加; 白色虚线框内分别表示缅甸安小叶蝉、额垠叶蝉族和外群等; 下同
Horizontal and vertical coordinates indicate the sequence numbers; color gradation in red indicates high correlation values; white dash line box indicate sequences ofAnakaburmensis, Mukariini and outgroup; the same below
1.Mukariamaculata; 2.Mukariaalbinotata; 3.Mukariabambusana; 4.Mohuniabifasciana; 5.Flatfrontapronga; 6.Tiaobeiniaemaiensis; 7.Xenovartaacuta; 8.Anatkinaillustris; 9.Anakaburmensi; 10.Malaxellaflava; 11.Malaxellatetracantha; 12.Meimunaopalifera

图6 COⅠ基因条形码矢量图Fig. 6 Klee-diagram for the COⅠ gene
1.Mukariabambusana; 2.Dryodurgadeslamellaris; 3.Mukariapallipes; 4.Mukariaalbinotata; 5.Mukariamaculata; 6.Olidianahuangmina; 7.Anakaburmensi; 8.Tiaobeiniaemaiensis; 9.Malaxellaflavai; 10.Malaxellatetracantha
Del-Prado R, Cubas P, Lumbsch HT,etal. 2010. Genetic distances within and among species in monophyletic lineages of Parmeliceae (Ascomycota) as a tool for taxon delimitation[J]. Molecular Phylogenetics and Evolution, 56: 125-133.
Dietrich CH, Whitcomb RF, Black IV WC. 1997. Phylogeny of the grassland leafhopper genusFlexamia(Homoptera: Cicadellidae) based on mitochondrial DNA sequences[J]. Molecular Phylogenetics and Evolution, 8: 139-149.
Fang QQ, Black IV WC, Blocker HD,etal. 1993. A phylogeny of New WorldDeltocephalus-like leafhopper genera based on mitochondrial 16S ribosomal DNA sequences[J]. Molecular Phylogenetics and Evolution, 2(2): 119-131.
Folmer O, Black M, Hoeh W,etal. 1994. DNA primers for amplification of mitochondrial cytochromecoxidase subunit Ⅰ from diverse metazoan invertebrates[J]. Molecular Marine Biology and Biotechnology, 3(5): 294.
Hajibabaei M, Janzen DH, Burns JM,etal. 2006. DNA barcodes distinguish species of tropical Lepidoptera[J]. Proceedings of the National Academy of Sciences of the United States of America, 103: 968-971.
Hebert PDN, Penton EH, Burns JM,etal. 2004a. Ten species in one: DNA barcoding reveals cryptic species in the neotropical skipper butterflyAstraptesfulgerator[J]. Proceedings of the National Academy of Sciences of the United States of America, 101: 14812-14817.
Hebert PDN, Ratnasingham S, Waard JR. 2003. Barcoding animal life: cytochrome c oxidase subunit Ⅰ divergences among closely related species[J]. Proceedings of the Royal Society B-Biological Sciences, 270: S96-S99.
Hebert PDN, Stoeckle MY, Zemlak TS,etal. 2004b. Identification of birds through DNA barcodes[J]. PLoS Biology,2: 1657-1663.
Köhler F. 2007. From DNA taxonomy to barcoding: how a vague idea evolved into a biosystematic tool[J]. Zoosystematics and Evolution, 83: 44-51.
Miglietta MP, Schuchert P, Cunningham CW. 2009. Reconciling genealogical and morphological species in a worldwide study of the family Hydractiniidae (Cnidaria, Hydrozoa)[J]. Zoologica Scripta, 38: 403-430.
Miranda LS, Collins AG, Marques AC. 2010. Molecules clarify a Cnidarian life cycle-the “Hydrozoan”Microhydrulalimopsicolais an early life stage of the StaurozoanHaliclystusantarcticus[J]. PLoS ONE, 5: e10182. DOI: 10.1371/journal.pone.0010182.
Moura CJ, Cunha MR, Porteiro FM,etal. 2011a. Polyphyly and cryptic diversity in the hydrozoan families Lafoeidae and Hebellidae (Cnidaria: Hydrozoa)[J]. Invertebrate Systematics, 25: 454-470.
Moura CJ, Cunha MR, Porteiro FM,etal. 2011b. The use of the DNA barcode gene 16S mRNA for the clarification of taxonomic problems within the family Sertulariidae (Cnidaria, Hydrozoa)[J]. Zoologica Scripta, 40: 520-537.
Ortman BD, Bucklin A, Pagès F,etal. 2010. DNA barcoding the Medusozoa using mtCOⅠ[J]. Deep Sea Research Part Ⅱ: Topical Studies in Oceanography, 57(24-26): 2148-2156.
Ratnasingham S, Hebert PDN. 2007. BOLD: the barcode of life data system (http://www.barcodinglife.org)[J]. Molecular Ecology Notes, 7: 355-364.
Schuchert P. 2006. The European athecate hydroids and their medusae (Hydrozoa, Cnidaria): Capitata Part 1[J]. Revue Suisse de Zoologie, 113: 325-410.
Sinniger F, Reimer JD, Pawlowski J. 2008. Potential of DNA sequences to identify zoanthids (Cnidaria: Zoantharia)[J]. Zoological Science, 25: 1253-1260.
Sirovich L, Stoeckle MY, Zhang Y. 2009. A scalable method for analysis and display of DNA sequences[J]. PLoS ONE, 4: e7051. DOI: 10.1371/journal.pone.0007051.
Sirovich L, Stoeckle MY, Zhang Y. 2010. Structural analysis of biodiversity[J]. PLoS ONE, 5: e9266. DOI: 10.1371/journal.pone.0009266.
Stampar SN, Maronna MM, Vermeij MJA,etal. 2012. Evolutionary diversification of banded tube-dwelling anemones (Cnidaria; Ceriantharia; Isarachnanthus) in the Atlantic Ocean[J]. PLoS ONE, 7: e41091. DOI: 10.1371/journal.pone.0041091.
Stoeckle MY, Coffran C. 2013. TreeParser-aided Klee diagrams display taxonomic clusters in DNA barcode and nuclear gene datasets[J]. Scientific Reports, 3: 2635-2637.
Sun W, Zhang ZT, Dong H,etal. 2013. Analysis of genetic differentiation and gene flow among different geographic populations ofOedaleusinfernalis(Orthoptera: Acrididae) based on mtDNA COⅠ gene sequences[J]. Acta Entomologica Sinica, 56(8): 907-916.
Sun Y, Li Q, Kong L,etal. 2012. DNA barcoding of Caenogastropoda along coast of China based on the COⅠ gene[J]. Molecular Ecology Resources, 12: 209-218.
Ward RD, Zemlak TS, Innes BH,etal. 2005. DNA barcoding Australia’s fish species[J]. Philosophical Transactions: Biological Sciences, 360: 1847-1857.
Yang L, Chen XS. 2011. Review of bamboo-feeding leafhopper genusMukariaDistant (Hemiptera: Cicadellidae: Mukariinae) with description of a new species from China[J]. Zootaxa, 2882: 27-34.
Yang ZH, Rannala B. 2010. Bayesian species delimitation using multilocus sequence data[J]. Proceedings of the National Academy of Sciences of the United States of America, 107: 9264-9269.
Zemlak TS, Ward RD, Connell AD,etal. 2009. DNA barcoding reveals overlooked marine fishes[J]. Molecular Ecology Resources, 9: 237-242.
Zhang JZ, Guo YP, Ma EB. 2004. DNA extraction and RAPD analysis of grasshopper samples kept under different conditions[J]. Chinese Journal of Zoology, 39(2): 53-57.
Zhang MZ, Kang L. 2001. Extraction of total DNA from locusts and optimization of reaction conditions for RAPD analysis[J]. Zoological Research, 22(1): 20-26.
猜你喜欢
河南师范大学学报(自然科学版)(2022年5期)2022-08-08
中国农业科学(2022年10期)2022-06-28
广东蚕业(2021年12期)2022-01-18
今日农业(2021年15期)2021-11-26
少年文艺·开心阅读作文(2021年8期)2021-09-05
中外葡萄与葡萄酒(2020年5期)2020-09-25
北京农学院学报(2019年3期)2019-10-14
小学科学(学生版)(2019年5期)2019-05-21
蔬菜(2018年5期)2018-05-17
学苑创造·B版(2017年10期)2017-12-21
