
非原位拉曼光谱区分富锂层状氧化物的集成结构:一种预测Li2MnO3活化程度的有效方法
2018-04-10 09:25陈丹丹李广社范建明李保云李国华李莉萍
无机化学学报 2018年4期
陈丹丹 李广社 范建明 李保云 张 丹 冯 涛 李国华 李莉萍*,
0 引 言
锂离子电池因具有高的能量密度、良好的循环性能、环境友好等优点而被广泛应用于便携电子设备和电动汽车领域[1-4]。而正极材料是限制锂离子电池容量和能量密度的瓶颈所在[5]。众多研究者致力于寻找高容量、高安全性和低成本的正极材料来替代目前主要的商业化LiCoO2材料[6-11]。富锂层状氧化物在首次充电过程中电压达到4.5 V以上时,Li2MnO3组分发生电化学活化贡献容量,从而使富锂材料显示出很高的容量(>250 mAh·g-1)[12-14]。同时,富锂材料因富含低成本的锰元素、安全性高等特点,被认为是最有前景的下一代锂离子电池正极材料[15-16]。然而,一些内在因素制约着富锂材料的容量和循环性能,进而制约着其商业化的发展。
一方面,由于Li+的半径与Ni2+的半径相近,材料中普遍存在Li/Ni混占现象[17]。Zhang等[18]发现采用不同原料合成的同组分富锂材料中Li/Ni混占程度存在差异。Zheng等[19]深入探究了Li/Ni混占对不同组分的层状材料热稳定性的影响。另一方面,在首次充放电过程中,Li2MnO3组分在4.5 V以上的活化充分与否直接影响富锂材料平台区容量的贡献,Li2MnO3的活化情况与富锂材料的两相集成方式有关。上述2方面的问题通常来源于合成条件的影响,不同的合成条件影响富锂材料的晶型、晶粒尺寸、相纯度、形貌、比表面积等方面,使得富锂材料的电化学性能与其对应的合成方法密切相关[20-21]。
富锂材料的两相集成方式目前公认的结论有2个,即分别用 Li1+(x/(2+x))M1-(x/(2+x))O2(M=Mn、Co、Ni、Cr等)表示的固溶体结构和x Li2MnO3·(1-x)LiMO2(M=Mn、Co、Ni、Cr等)表示的复合物结构,从结构角度分析,固溶体结构的富锂材料源于Li+在晶格中有序排列,不同种类的过渡金属离子随机地出现在晶格中的过渡金属位上[22]。复合物结构的富锂材料首先由Thackeray等[23-25]提出,并指出富锂材料由空间上有序分布的Li2MnO3域和无序分布的LiMO2域组成。根据两组分域的大小我们将复合物分为纳米复合物和非纳米复合物。在富锂材料两相集成方式与性能的关系的探究中,Gu等[26]发现复合结构的Li1.2Mn0.6Ni0.2O2晶粒表面和晶界处富镍而内部富锰的现象,即存在元素偏析现象。相比于在原子水平上均匀分布的固溶体结构的富锂材料,复合物结构的富锂材料多了晶粒、晶界和元素偏析这些因素的限制,这对于材料的电化学性能来说是不利的。此外,McCalla和Li等[27-28]通过结合电化学性能数据和XRD数据分析发现单相的固溶体结构的材料不具备纳米复合物材料对电化学性能带来的不利影响。Tang等[29]通过结合扫描透射电子显微镜和电化学性能分析发现在复合物结构的富锂材料内存在Li2MnO3域富集现象,使得材料在电化学循环过程中更不容易被活化而表现出差的电化学性能,即当富锂材料以固溶体结构存在或Li2MO3分离域的尺寸越小,在有限的充电过程中,材料活化的越充分,在4.5 V左右的电压平台越长,贡献的容量越多。目前探究富锂材料两相集成方式所采用的方法通常是高端的电镜技术,理论计算等难实现、难分析的技术表征手段 (如高角度环形暗场扫描透射电子显微镜(HAADF/STEM&D-STEM)、魔角旋转核磁共振(MASNMR)、X射线吸收光谱(XAS)等)和工艺复杂的电化学性能测试,而采用简单易行的方法探究富锂材料的两相集成方式,并将之与电化学性能相联系的研究十分缺乏。
基于此,我们采用5种方法,即溶胶-凝胶法、高温固相法、共沉淀法、水热法和溶剂热法合成了富锂锰基正极材料Li1.2Mn0.6Ni0.2O2。使用扫描电子显微镜(SEM)和X射线粉末衍射(XRD)对5个样品的形貌和体相结构进行了表征。更重要的是,我们采用非原位拉曼光谱对5个样品进行测试,并与文献中的Li2MnO3和LiMn0.5Ni0.5O2的拉曼光谱对比来分析样品的局部结构,通过着重分析5个样品在488 cm-1左右(ν1)和 600 cm-1左右(ν2)的拉曼峰确定两相集成方式。结合电化学性能测试结果,尤其是首次充电过程中由Li2MnO3活化贡献容量的多少来进一步探究材料的集成结构与电化学性能之间的联系。
1 实验部分
1.1 试剂与样品制备
本工作使用的试剂包括LiOH·H2O(纯度95%)、LiAc·4H2O(纯度 99%)、Ni(Ac)2·4H2O(纯度 98%)、Mn(Ac)2·4H2O(纯度 99%)、一水合柠檬酸(纯度 99.5%)、NaOH(片状,纯度 96%)、尿素(纯度 99%)。
采用5种合成方法,即溶胶-凝胶法、高温固相法、共沉淀法、水热法和溶剂热法合成Li1.2Mn0.6Ni0.2O2正极材料。
(1) 溶胶-凝胶法
所采取的溶胶-凝胶法属溶胶-凝胶机制中的配合物型。 LiAc·2H2O、Mn(Ac)2·4H2O 和 Ni(Ac)2·4H2O 按 照 nLi∶nMn∶nNi=6∶3∶1 溶于 200 mL 去离 子 水中,柠檬酸作为配位剂,浓氨水调节溶液pH值在7~8之间得到金属离子配合物溶胶,置于80℃的水浴中蒸干形成金属离子配合物凝胶。得到的凝胶于180℃的烘箱中发泡一夜并充分研磨后置于马弗炉中500℃保温5 h,再于900℃下反应12 h。得到的样品记为SG。
(2) 高温固相法
以金属乙酸盐为原料,按照化学计量比投料并充分研磨混合均匀后置于马弗炉内500℃热处理6 h后取出进行二次研磨,二次研磨后的粉体置于马弗炉内900℃煅烧12 h,得到的样品记为HS。
(3) 共沉淀法
以过渡金属硫酸盐为原料,先按化学计量比将过渡金属硫酸盐溶于去离子水中,配成体积为100 mL的溶液A。再配制20 mL 2 mol·L-1的NaOH溶液与10 mL浓氨水溶于70 mL去离子水中配成等体积的溶液B。溶液A与溶液B在Ar气氛下,同时滴加进反应容器进行共沉淀反应,滴加速率为1 drop·s-1。滴加完毕后搅拌一夜令其沉淀完全,抽滤,用去离子水洗涤除去残留的Na+等杂质元素后于80℃烘箱中烘干得到前驱体,将其与锂源混合,研磨均匀于900℃马弗炉中煅烧反应12 h,得到的样品记为CP。
(4) 水热法
以过渡金属乙酸盐和尿素为原料,按照过渡金属与尿素的物质的量比为1∶2投料于100 mL反应釜内,加入60 mL去离子水配成均一澄清的溶液,置于180℃烘箱中反应15 h后,对产物抽滤,洗涤,烘干,得到前驱体。将前驱体与锂源混合,充分研磨后于马弗炉内900℃煅烧12 h,得到的样品记为HT。
(5) 溶剂热法
以过渡金属乙酸盐和尿素为原料,按照过渡金属与尿素的物质的量比为1∶1.2投料于100 mL反应釜中,以20 mL无水乙醇与40 mL去离子水的混合溶液作为反应介质配成澄清均一的溶液,于200℃烘箱中反应36 h,对反应产物抽滤,洗涤,烘干,得到前驱体。将前驱体与锂源充分研磨混合均匀于900℃马弗炉内反应12 h,得到的样品记为ST。
1.2 样品表征
X射线衍射(XRD)测试采用日本Rigaku公司MiniFlex600型 X射线衍射仪,Cu Kα 射线(λ=0.154 18 nm),管电流 15 mA,管电压 40 kV,扫描范围 10°~80°,扫描速度 2°·min-1,步长 0.02 °,光谱纯KCl作为内标进行衍射峰位的校正;样品形貌观察采用日本JEOL公司的JSM-6700型场发射扫描电子显微镜(SEM),加速电压为 10 kV,分辨率为 1 μm;采用法国Jobin Yvon公司的JY-HR800型激光拉曼光谱仪对样品进行拉曼光谱测试,激发波长为532 nm,测试的波数范围为200~800 cm-1;采用法国Jobin Yvon公司的Ultima 2型电感耦合等离子体原子发射光谱(ICP-AES)对样品中的元素组成进行分析。
1.3 电池组装与性能测试
将合成的电极材料、导电碳黑和PVDF以质量比8∶1∶1与一定量N-甲基吡咯烷酮溶剂混合制成浆料均匀地涂在铝箔上,于100℃真空烘箱烘干12 h,活性物质质量为2~2.5 mg。以金属锂片为对电极,1 mol·L-1LiPF6/(碳酸乙烯酯+碳酸二甲酯+碳酸甲乙酯)(LiPF6/(EC+DMC+EMC))(VEC∶VDMC∶VEMC=1∶1∶1)为电解液,聚丙烯材料为隔膜,在充满氩气的手套箱内组装成2025型扣式电池。在深圳新威尔电子有限公司Neware测试系统上进行电化学性能测试,充放电测试的电压区间为2.0~4.8 V。
2 结果与讨论
图1为5个样品的扫描电镜(SEM)图,可以看出5个样品形貌类似,都是不规则的颗粒,存在部分团聚现象,样品HT和ST较其他3个样品团聚的更严重一些。样品SG、HS和CP尺寸较小,约为100~400 nm,样品HT和ST的颗粒粒径分布散乱,在100 nm到1μm之间都有所分布。因5个样品形貌相近,均为无规则颗粒,因此形貌不是影响电化学性能的主要因素。
图2为5个富锂材料的X射线衍射(XRD)图,可以明显地看出5种方法所合成的样品均具有典型的六方晶系(空间群为R3m)层状结构[30]。在38°和65°左右分别对应(006)和(012)、(018)和(110)两对清晰可见的劈裂峰,是层状材料的特征衍射峰[4]。在20°~25°之间存在一些微弱的衍射峰与单斜晶系C2/m空间群的超晶格结构密切相关,这是由过渡金属层中独特的LiMn6有序结构贡献的[31]。从图2中给出的20°~25°的XRD放大图我们可以看出,归属于C2/m空间群的3个晶面(020)、(110)和(110)的衍射峰强度随着合成方法的不同而不同。表1给出了XRD精修拟合的结构参数,c/a值可以代表三方畸变的程度,当c/a大于4.899时证明材料中的阳离子高度有序[20],5个样品的c/a值相近,具有相近的阳离子有序结构。Li/Ni disordering代表Li/Ni混占程度,5个样品的Li/Ni混占程度相近。同时,5个样品的XRD拟合误差Rwp、Rp和χ2相近,故5个富锂样品的体相结构基本相同,不存在明显差别。

图1 样品的SEM图Fig.1 SEM images of as-prepared samples

图2 样品的XRD图Fig.2 XRD patterns of as-prepared samples
样品整体的晶体结构信息可通过多种衍射方法来获得,但是针对关系到富锂材料电化学性能的过渡金属离子周围的局部结构采用拉曼光谱来分析更加地简单高效[32-34]。图3(a)是样品的拉曼光谱图,图3(b)是从文献中引用的Li2MnO3及LiNi0.5Mn0.5O2的拉曼光谱图[35-36],对比二者旨在找到5个同组分的富锂样品在局部结构上的区别。Li2MnO3具有 Ag(621、574、442、252 cm-1)和 Bg(502、422、377、330 cm-1)8个拉曼振动模式[35]。LiNi0.5Mn0.5O2具有2个拉曼振动模式,分别为600 cm-1的A1g振动模和500 cm-1左右以宽峰形式存在的Eg振动模[36-38]。由于富锂材料在结构上存在争议,在分析5个样品的拉曼光谱时做出如下假设,即富锂材料的拉曼光谱是由各组分的局部组分相叠加的结果。Li2MnO3和LiNi0.5Mn0.5O2的拉曼光谱发生叠加作用的区域在450~700 cm-1之间。小于450 cm-1部分的拉曼振动全部来自Li2MnO3组分,这也是富锂材料相比于LiMO2(M=Mn、Co、Ni)的特征峰。样品 SG、HS、HT 和ST小于450 cm-1的几个峰很明显,由此说明材料中存在类似于Li2MnO3的局部对称性。然而样品CP除了425 cm-1存在一个比较弱的峰之外,其他的峰并不明显,证明样品CP中并不存在明显的Li2MnO3域。根据峰位置分析5个样品在488 cm-1左右(ν1)和 600 cm-1左右(ν2)的拉曼峰由 Li2MnO3和LiNi0.5Mn0.5O2共同贡献。 ν1峰是由 Li2MnO3的 Bg(502 cm-1)和 LiNi0.5Mn0.5O2的 Eg(503 cm-1)共同贡献。ν2峰是由 Li2MnO3的 Ag(621 cm-1)和 LiNi0.5Mn0.5O2的A1g(600 cm-1)共同贡献。表2给出了5个样品2个拉曼峰ν1和ν2的具体参数。

表1 样品精修后的结构参数Table 1 Structural parameters for all samples refined by GSASsoftware
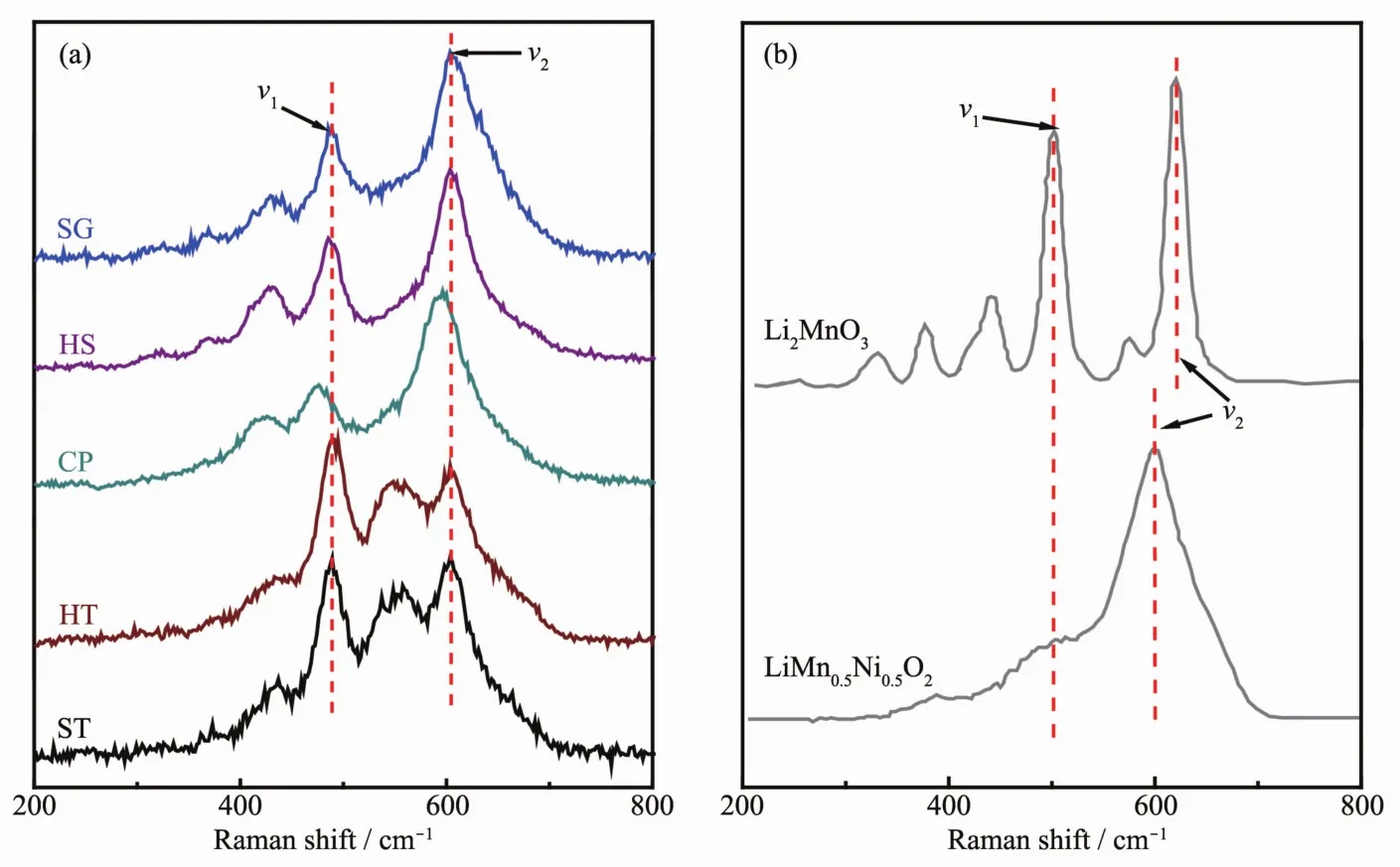
图3 (a)5个样品的拉曼光谱图;(b)Li2MnO3和LiNi0.5Mn0.5O2的拉曼光谱[35-36]Fig.3 Raman spectra of as-prepared samples(a),Li2MnO3 and LiNi0.5Mn0.5O2(b)
五个样品的 ν1峰相较于 Li2MnO3的 Bg(502 cm-1)和LiNi0.5Mn0.5O2的Eg(503 cm-1)均向低波数方向移动,这是因为Li2MnO3的Bg(502 cm-1)和LiNi0.5Mn0.5O2的Eg(503 cm-1)相重合,由于二者峰形差别较大使得ν1峰位发生明显的移动,但我们发现样品CP移动的幅度要比其他4个样品大一些。样品SG、HS、HT和 ST的 ν2峰相较于 LiNi0.5Mn0.5O2的 A1g振动模式轻微地向高波数方向移动,即向着Li2MnO3的Ag振动模方向移动,这是材料中存在的Li2MnO3域造成的,是两者相互叠加的结果,而两组分叠加并不能解释样品CP的ν1峰向低波数方向移动这种情况。最值得注意的是在550~575 cm-1处的拉曼振动,此处的振动峰来源于Li2MnO3的Ag(574 cm-1)。可以明显地看到在样品SG、HS、CP中此处的拉曼峰强度较弱,峰形不明显,与ν2峰以类似于肩峰的形式存在。而样品HT和ST在此处的拉曼振动峰比较尖锐,具有明显的峰形,与ν2峰以类似于劈裂峰的形式存在。我们可以看到Li2MnO3的Ag(574 cm-1)就很明显和尖锐,由此可以说明,样品HT和ST中存在较大的Li2MnO3域。比较5个样品ν2和ν1的峰强度 Iν2/Iν1, 样品 SG 和 HS 的 Iν2/Iν1相近, 且均与LiNi0.5Mn0.5O2接近, 样品 HT 和 ST 的 Iν2/Iν1相近,并靠近 Li2MnO3的 Iν2/Iν1。 而样品 CP 与其他样品显著不同, 其 Iν2/Iν1要比Li2MnO3和LiNi0.5Mn0.5O2的都要大很多。综上所述,采用由Li2MnO3和LiNi0.5Mn0.5O2共同贡献的假设并不能得到很好的解释样品CP的拉曼谱图,所以样品CP很有可能是以固溶体结构存在的;样品HT和ST的拉曼振动峰高度表现出了Li2MnO3的振动特征,二者可能是以复合物结构存在;样品SG和HS的拉曼光谱介于上述2种类型之间,即以Li2MnO3和LiNi0.5Mn0.5O2共生的纳米复合结构存在。
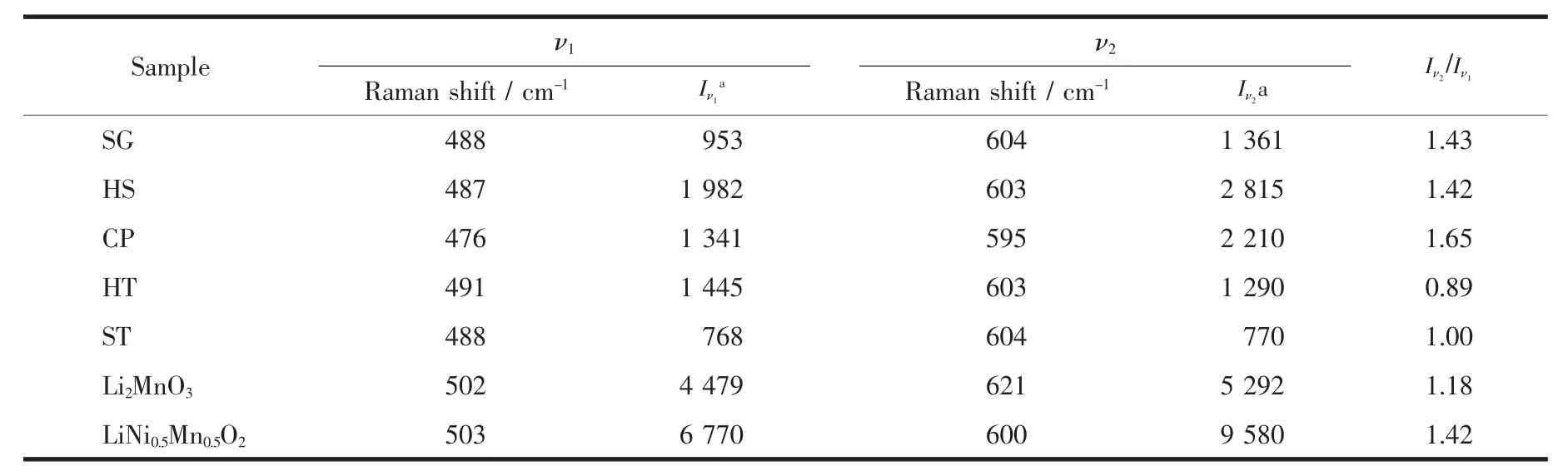
表2 样品的特征拉曼振动峰相关数据Table 2 Raman band data for all samples
通过结合前人的工作探究和本文中我们通过拉曼光谱分析得到的5个富锂样品的两相集成方式信息,我们预测具有固溶体结构的样品CP电化学性能可能最优,其次是具有纳米复合物结构的样品SG和HS,而具有非纳米复合物结构的样品HT和ST的电化学性能有可能最差。
采用ICP-AES对样品的Li、Mn、Ni元素组成进行分析,在相同金属投料比的条件下,采用不同方法合成的样品中,金属元素组成相近。5个样品均于室温条件下2.0~4.8 V(vs Li+/Li)的电压范围内进行恒电流充放电的电化学性能测试。图4(a)为5个样品在20 mA·g-1下的首次充放电曲线。样品SG、HS和CP具有相似的首次充放电曲线,即在首次充电过程中存在明显的2个阶段,第一个阶段是位于3.8~4.5 V的斜坡区域,这是由LiNi0.5Mn0.5O2中Li+脱出伴随着Ni2+被氧化成Ni4+所贡献的。第2个阶段为位于4.5 V左右的电压平台,这部分容量来源于Li2MnO3活化,脱出Li2O而形成类MnO2相[18]。样品HT和ST的首次充放电曲线明显不同于样品SG、HS和CP,其由Li2MnO3活化贡献的容量平台很短,几乎看不到。表3给出了5个样品各个阶段理论充电比容量和在20 mA·g-1下实际充电比容量及二者之间的差值。对比5个样品的斜坡区域,发现样品HT和ST的ΔCLiNi0.5Mn0.5O2最大,其后依次是样品CP、HS和SG。而样品SG和HS的在这一阶段的实际充电比容量要高于理论充电比容量,这种情况的出现源于多种因素的叠加,如组分的细微差异、测试系统误差,活性物质测量带来的误差都会造成实际值高于理论值,因此这一部分容量的损失我们不能够用准确的某一个因素去判断,需要综合多种因素进行考量。我们认为理论比容量与实际比容量的差值在±10 mA·g-1左右都是正常。本文中我们主要探究两相集成方式对Li2MnO3活化情况的影响,关于第一阶段的影响因素不再进行深入探讨。根据表3,在首次充电过程中, 样品 CP、SG、HS、ST和 HT的分别为 47.88、61.84、73.14、167.93 和 169.28 mAh·g-1。样品CP由Li2MnO3活化贡献的实际充电比容量最接近理论充电比容量,Li2MnO3更容易被活化。相应地,样品ST和HT中的Li2MnO3很难被活化,表现出很低的实际充电比容量。样品SG和HS的容量介于上述三者之间。图4(b)给出了5个样品于100 mA·g-1下的循环性能图。经历100次循环后,样品SG、HS、CP、HT和ST的放电比容量由首次的 放 电 比 容 量 203.8、163.4、199.2、50.6 和 65.3 mAh·g-1变为 173.7、151.8、185.3、55.3 和 101.8 mAh·g-1,容量保持率分别为 85.3%、93.3%、93.0%、107.8%和156.9%。由此可见,样品HT和ST中的Li2MnO3在首次循环过程中没有得到充分活化,在后续循环过程中缓慢活化,使得放电比容量在后续的循环中逐步上升,再次证明了在样品HT和ST中的Li2MnO3组分在首次充电过程中难以活化这个事实。

图4 样品(a)在20 mA·g-1下的首次充放电曲线和(b)在100 mA·g-1下的循环性能曲线Fig.4 (a)First charge-discharge curves at a current density of 20 mA·g-1 and(b)cycling performances at a current density of 100 mA·g-1 of as-prepared samples
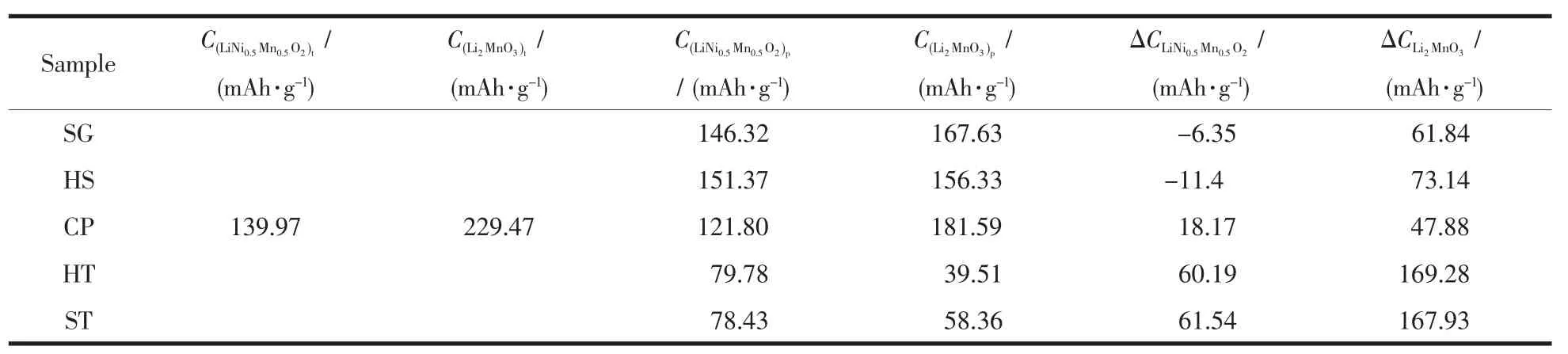
表3 样品中2个组分通过计算得到的理论放电比容量C t和电化学性能测试得到的实际放电比容量C p及其二者的差值ΔCTable 3 A comparison of theoretical first charge capacities(C t)by each component,practical charge capacities(C p)obtained by electrochemical tests and deviation of theoretical capacities and pratical capacities(ΔC)
图5为不同电流密度下5个样品的首次充放电曲线。样品HT和ST在不同电流密度下的首次充电曲线在4.5 V左右的平台区域仍旧不明显。同时,从图5中可以看出样品SG、HS和CP具有更好的倍率性能,尤其是样品CP。在20、100、200和400 mA·g-1电流密度下,样品CP首次放电比容量分别为 218.0、199.2、193.7 和 178.7 mAh·g-1。 证 明 了Li2MnO3组分活化为材料贡献了高的充电比容量,使其在放电过程中也呈现出高的比容量。

图5 五个样品在不同电流密度下的首次充放电曲线Fig.5 First charge-discharge curves at different current density
图 6为 5个样品在 100、200和 400 mA·g-1下的循环性能图。可以看到,在200 mA·g-1下,样品SG、HS、CP、HT和 ST经历 100次循环后放电比容量分别为 154.2、138.7、164.1、45.6 和 61.5 mAh·g-1,容量保持率分别为80.0%、88.4%、84.7%、108.6%和117.6%。 在 400 mA·g-1下,样品 SG、HS、CP、HT 和ST经历100次循环后放电比容量分别为144.5、124.2、169.7、43.6 和 59.7 mAh·g-1,容量衰减率分别为17.5%、15.3%、5.00%、-11.4%和-18.3%。结合对图4(b)和图5的分析,我们发现样品HT和ST的循环性能的反常现象在其他电流密度下仍旧存在,再次说明了样品HT和ST中的Li2MnO3在电化学循环过程中难以被活化。
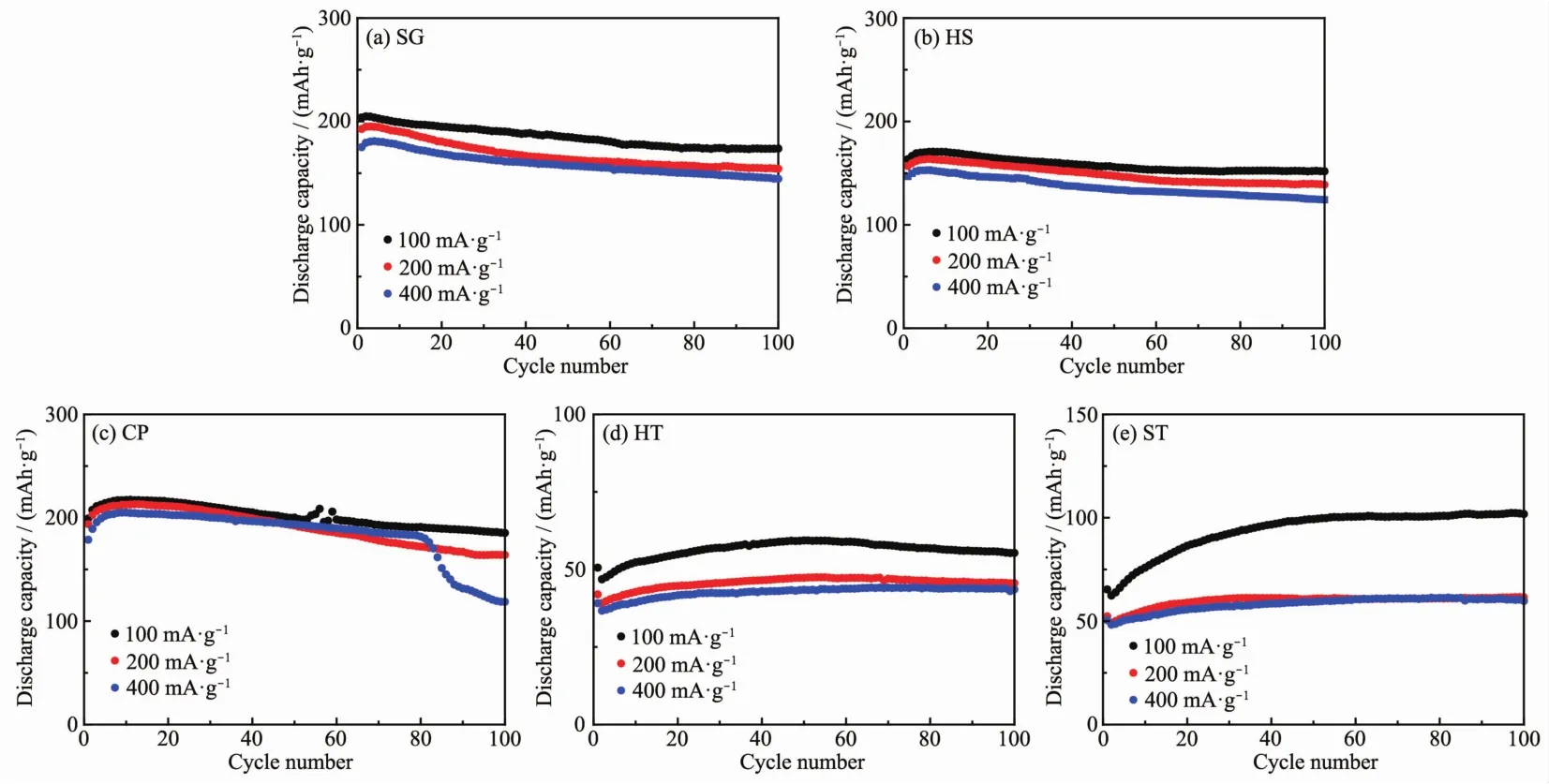
图6 五个样品在不同电流密度下的循环性能曲线Fig.6 Cycling performances of as-prepared samples at different current density
3 结 论
本文通过5种方法,即溶胶-凝胶法、高温固相法、共沉淀法、水热法和溶剂热法合成了Li1.2Mn0.6Ni0.2O2正极材料。SEM和XRD分析表明5个富锂样品形貌相近,体相结构相同。拉曼光谱测试分析发现5个样品的两相集成方式不同,样品CP倾向于以固溶体结构存在,样品SG和HS以纳米复合物形式存在,样品HT和ST以非纳米复合物形式存在。结合前人工作我们对5个样品的电化学性能进行了预测,即在首次充电过程中样品CP中的Li2MnO3可以充分活化使其具有较高的容量,较好的电化学性能,其次是样品SG和HS,最差的是样品HT和ST。通过电化学性能测试发现样品CP的ΔCLi2MnO3为 47.88 mAh·g-1,由 Li2MnO3活化贡献的实际充电比容量最接近理论充电比容量,Li2MnO3更容易被活化。样品ST和HT的分别为167.93 和 169.28 mAh·g-1, 其中的 Li2MnO3很难被活化,表现出很低的实际充电比容量。这与我们预测的电化学性能结果相吻合,由此我们认为可以通过非原位拉曼光谱测试来区分富锂材料的集成结构进而预测富锂材料的电化学性能。
参考文献:
[1]Choi N S,Chen Z,Freunberger S A,et al.Angew.Chem.Int.Ed.,2012,51(40):9994-10024
[2]Santhanam R,Rambabu B.J.Power Sources,2010,195(17):5442-5451
[3]Dunn B,Kamath H,Tarascon JM.Science,2011,334(6058):928-935
[4]Zhang X H,Luo D,Li G S,et al.J.Mater.Chem.A,2013,1(34):9721-9729
[5]Goodenough J B,Kim Y.Chem.Mater.,2010,22(3):587-603
[6]Yu H,Ishikawa R,So Y G,et al.Angew.Chem.Int.Ed.,2013,52(23):5969-5973
[7]Zhao X,Reddy M V,Liu H,et al.RSC Adv.,2014,4(47):24538-24543
[8]Yu H,Kim H,Wang Y,et al.Phys.Chem.Chem.Phys.,2012,14(18):6584-6595
[9]WANGHai-Yan(王海燕),TANG Ai-Dong(唐爱东),HUANG Ke-Long(黄可龙),et al.Chinese J.Inorg.Chem.(无机化学学报),2008,24(4):593-599
[10]Wei WF,Chen L B,Pan A Q,et al.Nano Energy,2016,30:580-662
[11]WEI Xuan-Ni(韦旋妮),LAI Qiong-Jie(赖琼钰),GAO Yuan(高媛),et al.Chinese J.Inorg.Chem.(无机化学学报),2005,21(7):999-1003
[12]Ohzuku T,Nagayama M,Tsuji K,et al.J.Mater.Chem.,2011,21(27):10179-10188
[13]Thackeray M M,Kang S H,Johnson C S,et al.J.Mater.Chem.,2007,17(30):3112-3125
[14]Song B,Lai M O,Liu Z,et al.J.Mater.Chem.A,2013,1(34):9954-9965
[15]Lee S,Kim E Y,Lee H,et al.J.Power Sources,2014,269:418-423
[16]JIANG Yao-Xue(姜摇雪),SHI Nan-Nan(史楠楠),ZHANG Yao-Ying(张摇莹),et al.Chem.J.Chinese Universities(高等学校化学学报),2015,36(4):739-744
[17]Dixit H,Zhou W,Idrobo J,et al.ACS Nano,2014,8(12):12710-12716
[18]Zhang X H,Yu C,Huang X D,et al.Electrochim.Acta,2012,81(30):233-238
[19]Zheng J X,Liu T C,Hu Z X,et al.J.Am.Chem.Soc.,2016,138(40):13326-13334
[20]Ben-Kamel K,Amdouni N,Mauger A,et al.J.Alloys Compd.,2012,528(5):91-98
[21]ZHENG Zhuo(郑卓),HUA Wei-Bo(滑纬博),WU Zhen-Guo(吴振国),et al.Chinese J.Inorg.Chem.(无机化学学报),2017,33(2):307-314
[22]Lu Z H,Chen Z H,Dahn JR,et al.Chem.Mater.,2013,15(16):3214-3220
[23]Johnson CS,Kim JS,Lefief C,et al.Electrochem.Commun.,2004,6(10):1085-1091
[24]Thackeray M M,Johnson C S,Vaughey JT,et al.J.Mater.Chem.,2005,15(23):2257-2267
[25]Thackeray M M,Kang S H,Johnson C S,et al.J.Mater.Chem.,2007,17(30):3112-3125
[26]Gu M,Genc A,Belharouak I,et al.Chem.Mater.,2013,25(11):2319-2326
[27]McCalla E,Li J,Rowe A W,et al.J.Electrochem.Soc.,2014,161(4):A606-A613
[28]Li J,Camardese J,Glazier S,et al.Chem.Mater.,2014,26(24):7059-7066
[29]Tang D C,Liu D T,Liu Y Y,et al.Prog.Nat.Sci.,2014,24(4):388-396
[30]Fan J M,Li G S,Li G H,et al.Electrochim.Acta,2017,245(10):118-127
[31]Jarvis K A,Deng Z Q,Allard L F,et al.Chem.Mater.,2011,23(16):3614-3621
[32]Inaba M,Iriyama Y,Ogumi Z,et al.J.Raman Spectrosc.,1997,28(8):613-617
[33]Ammundsen B,Burns GR,Islam M S,et al.J.Phys.Chem.B,1999,103(25):5175-5180
[34]Hwang SJ,Park H S,Choy J H,et al.Electrochem.Solid-State Lett.,2001,4(12):A213-A216
[35]Ruther R E,Dixit H,Pezeshki A M,et al.J.Phys.Chem.C,2015,119(32):18022-18029
[36]Karan N K,Saavedra-Arias J J,Pradhan D K,et al.Electrochem.Solid-State Lett.,2008,11(8):A135-A139
[37]Jeong SK,Song C H,Nahm K S,et al.Electrochim.Acta,2006,52(3):885-891
[38]Julien C.Solid State Ionics,2000,136-137(2):887-896
猜你喜欢
辽宁省博物馆馆刊(2021年0期)2021-07-23
中学生数理化(高中版.高考理化)(2021年4期)2021-07-19
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
表面工程与再制造(2019年6期)2019-08-24
中国粮油学报(2018年12期)2018-03-19
资源节约与环保(2018年1期)2018-02-08
原子与分子物理学报(2015年3期)2015-11-24
原子与分子物理学报(2015年1期)2015-11-24
国外科技新书评介(2014年8期)2014-12-05
