
CDK4/6抑制剂abemaciclib合成工艺的优化
2018-05-03 05:33冯权武尹传奇
武汉工程大学学报 2018年2期
鄢 龙,傅 晶,吴 莉,冯权武,尹传奇
武汉工程大学化学与环境工程学院,湖北 武汉 430205
Abemaciclib,化学名为 N-[5-[(4-乙基-1-哌嗪)甲基]2-吡啶基]-5-氟-4-[4-氟-1-异丙基-2-甲基-1H-苯并咪唑-6-基]-2-嘧啶胺。作为细胞周期蛋白依赖性激酶(cyclin-dependent kinase,CDK)4/6特异性抑制剂,abemaciclib对乳腺癌HER2阴性的晚期或复发乳腺癌的治疗获得了美国食品药物管理局(FDA)“突破性药物”认证[1-2]。2017年9月28日,FDA批准礼来公司的乳腺癌新药abemaci⁃clib(商品名 Verzenio)上市[3],因而对该药物合成工艺的探索具有很大的应用价值。
Abemaciclib的合成路线[4-5]主要包括路线1以6-氨基烟酸乙酯和3,5-二氟-4-氨基苯乙酮为原料,经十一步反应合成,总收率是11.0%。该路线在反应过程中使用LiAlH4,具一定的危险性,工艺过于繁琐,工业化所需的成本高,不利于工业化生产;路线2以6-溴-3-吡啶甲醛和2,6-二氟苯胺为原料,经8步反应合成,总收率是15.4%。该路线在反应过程中使用NaH,具一定的危险性,不易于工业化生产;路线3以4-溴-2,6-二氟苯胺为原料合成中间体 6-(2-氯-5-氟嘧啶-4-基)-4-氟-1-异丙基-2-甲基-1H-苯并[d]咪唑,该中间体继续与6-氨基-3-吡啶甲醛和N-乙基哌嗪反应合成abemaci⁃clib,总收率为20.0%。但中间体与N-乙基哌嗪易发生副反应,分离困难;路线4以N-乙基哌嗪和6-溴-3-吡啶甲醛为原料反应合成中间体5-((4-乙基哌嗪-1-基)甲基吡啶-2-胺,再与路线3的中间体 6-(2-氯-5-氟嘧啶-4-基)-4-氟-1-异丙基-2-甲基-1H-苯并[d]咪唑偶联合成abemaciclib,总收率为25.0%(见图1)。
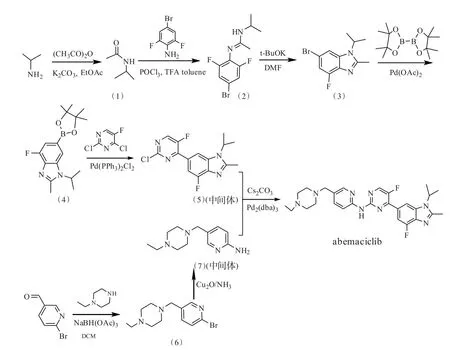
图1 abemaciclib的合成路线Fig.1 Synthetic route of abemaciclib
综上所述,考虑到反应辅料的安全性及易于工业化的要求,本研究选择在合成路线4的基础上,对相关合成和分离条件进行优化,使反应条件温和、产物易分离纯化和收率更高,易于工业化生产。
1 实验部分
1.1 试剂与仪器
所用试剂均为国产市售分析纯。氮气(体积分数98%)(武汉祥云公司)。
RY-1型熔点测试仪(天津天光光学仪器有限公司);DPX 300核磁共振仪(德国Bruker公司);5975C型气-质联用仪(美国安捷伦科技有限公司);LC-20AT型高效液相色谱仪(HPLC,日本岛津公司)。
检测方法:每次取10 mg,用甲醇稀释到需要浓度。然后用一个干净的注射器连接一个0.22 μm的筒式过滤器过滤样品进样。流速为2 mL/min,测量波长为254 nm。梯度洗脱。
1.2 实验过程
1.2.1 异丙基乙酰胺(1)的合成 先将85.0 mL(1.0 mol)异丙胺、138.4 mL(1.0 mol)三乙胺和200 mL乙酸乙酯,加入至1 000 mL三颈瓶中,搅拌降温至0 ℃,滴加89.7 mL(0.95 mol)乙酸酐,温度控制在(0±5)℃。滴完后,让其自然回复至室温,搅拌过夜。滴加约100 mL水,控制温度20℃以下,然后用饱和碳酸氢钠水溶液调节pH至8左右,分层,水相用乙酸乙酯萃取(100 mL×3),合并有机相,无水Na2SO4干燥,浓缩得无色澄清液体94.7 g,收率93.8%。ESI-MS m/z:101.2。
1.2.2 (E)-N'-(4-溴-2,6-二氟苯基)-N-异丙基乙脒(2)的合成
将 94.7 g(0.94 mol)化合物 1、900 mL 甲苯 、161.5 g(0.78 mol)4-溴-2,6-二氟苯胺和 162.6 mL(1.18 mol)三乙胺,加入至2 000 mL三颈瓶中水浴搅拌,滴加 110.0 mL(1.18 mol)三氯氧磷,控制温度10℃~30℃。滴加完后,水浴搅拌30 min,然后缓慢升温至110℃,回流反应2 h。降温至室温,滴加约100 mL饱和碳酸氢钠水溶液,然后减压浓缩除去甲苯,在得到的固液混合物中加入800 mL乙酸乙酯,搅拌10 min,再用饱和碳酸钠水溶液调pH至8左右,固体在调pH过程中会慢慢溶解至溶液澄清,静置分液,水相用乙酸乙酯萃取1次,合并有机相,无水Na2SO4干燥,浓缩得米白色固体227.0 g,收率100%。熔点:112℃。1H NMR(400 MHz,CDCl3)δ 7.02(d,J=5.9 Hz,2H),4.17(s,1H),1.74(s,3H),1.41(d,J=6.9 Hz,1H),1.22(d,J=6.2 Hz,16H)。
1.2.3 6-溴-4-氟-1-异丙基-2-甲基-1H-苯并[d]咪唑(3)的合成 将 227.0 g(0.78 mol)化合物 2、2 000 mL N,N'-二甲基甲酰胺和 150.0 g(1.56 mol)叔丁醇钠,加入至5 000 mL三颈瓶中,升温90℃~100℃加热搅拌,反应4 h~5 h。将反应冷却至室温,倒入6 000 mL水中,乙酸乙酯萃取,合并有机相,水洗2次,饱和食盐水洗1次,无水Na2SO4干燥,浓缩得米白色固体。将粗品加入100 mL v(甲叔醚)/v(石油醚)(1/10)溶液中,搅拌打浆30 min,过滤,然后用20 mL v(甲叔醚)/v(石油醚)(1/10)淋洗,干燥得米白色固体176.0g,收率83.0%。熔点:104℃。ESI-MS m/z:271.05。1H NMR(400 MHz,CDCl3)δ 7.37(s,1H),7.02(d,J=9.7 Hz,1H),4.58(m,J=14.0-6.9 Hz,1H),2.57(s,3H),1.57(d,J=6.9 Hz,6H)。
1.2.4 4-氟-1-异丙基-2-甲基-6-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)-1H-苯并[d]咪唑(4)的合成 将 176.0 g(0.65 mol)化合物 3、247.8 g(0.98 mol)双 联 硼 酸 频 哪 醇 酯 、191.1 g(1.95 mol)乙酸钾和2 000 mL N,N'-二甲基甲酰胺加入至5 000 mL三颈瓶中,氮气置换3次,快速加入17.6 g(25.10 mmol)双(三苯基膦)二氯化钯,然后搅拌升温至90℃。反应30 min后,取样,HPLC跟踪监测原料反应完全,将反应降温至室温,倒入4倍体积水中,乙酸乙酯萃取,有机相用硅藻土过滤1次,然后水洗2次,饱和食盐水洗1次,无水Na2SO4干燥,浓缩得304.8 g黑色固体,收率>100%(按收率100%计,可直接用于下步反应)。
1.2.5 6-(2-氯-5-氟嘧啶-4-基)-4-氟-1-异丙基-2-甲基-1H-苯并[d]咪唑(5)的合成 在5 000 mL三颈瓶中,将137.8 g(1.3 mol)碳酸钠溶于500 mL水中,加入1 000 mL乙二醇二甲醚和129.5 g(0.78 mol)2,4-二氯-5-氟嘧啶,氮气置换3次,快速加入20.3 g(29.00 mmol)双(三苯基膦)二氯化钯,升温至(80±5)℃,搅拌下滴加206.7 g(0.65 mol)化合物4溶于700 mL乙二醇二甲醚的溶液,滴完后于(80±5)℃搅拌反应2 h。将反应降温至室温,加入2 000 mL水,搅拌1 h,过滤用水淋洗,得灰色固体。将灰色固体加到200 mL异丙醇中,室温打浆搅拌30 min,过滤,少量异丙醇淋洗,抽干得灰白色固体144.3 g,收率68.9%。熔点:192℃。ESI-MS m/z:323.1[M+H]+。
1.2.6 1-((6-溴吡啶-3-基)甲基)-4-乙基哌嗪(6)的合成 将78.6 mL(0.62 mol)N-乙基哌嗪、98.1 g(0.53 mol)6-溴-3-吡啶甲醛、1 000 mL二氯甲烷和4.0 mL(0.07 mol)乙酸,加入至2 000 mL三颈瓶中,降温至0℃搅拌,分批加入160.0 g(0.74 mol)三乙酰基硼氢化钠,控制温度0℃~10℃,反应5 h。反应完全后,滴加2 mol/mL氢氧化钠水溶液调节pH至11左右,分层,水相用二氯甲烷萃取3次,合并有机相,水洗1次,无水Na2SO4干燥,浓缩得到150.0 g油状物,收率 100%。ESI-MS m/z:283.07[M+H]+。
1.2.7 5-((4-乙基哌嗪-1-基)甲基)吡啶-2-胺(7)的合成 将150.0 g(0.53 mol)化合物6、500 mL乙二 醇 、3.8 g(26.5 mmol)氧 化 亚 铜 、与 14.35 g(0.11 mol)碳酸钾、1 600 mL(10.6mol)氨水和18.7 mL(0.17 mol)N,N'-二甲基乙二胺(DMEDA)加入至5 000 mL三颈瓶中,搅拌,升温至60℃反应5 h。降温至室温,加入700 mL饱和NaCl水溶液,用配制好的6 mol/mL的NaOH溶液调节pH至12,用二氯甲烷萃取2次,无水Na2SO4干燥有机相,浓缩,将粗品加入到150 mL甲基叔丁基醚中,室温下搅拌。抽滤,用甲基叔丁基醚淋洗滤饼,得到白色固体99.1 g,收率90.0%。熔点:83℃。ESI-MS m/z:221.19[M+H]+。1H NMR(400 MHz,CDCl3)δ 7.92(s,1H),7.85(s,2H),7.30(m,2H),6.40(d,J=16.1,8.5 Hz,1H),3.40(s,2H),2.61-2.02(m,10H),0.98(t,J=7.1 Hz,3H)。
1.2.8 abemaciclib的合成 将144.3 g(0.45 mol)化合物 5、1 242.0 g(9.0 mol)碳酸钾、12.4 g(0.09 mol)4,5-双(二苯基膦)-9,9-二甲基氧杂蒽、700 mL叔戊醇和99.1 g(0.45 mol)化合物7加入至2 000 mL三颈瓶中,氮气置换3次,快速加入14.4 g(15.72 mmol)三(二亚苄基丙酮)二钯,搅拌,升温至102℃回流反应5 h,溶液呈棕黄色。将反应冷却至室温,加入150 mL二氯甲烷稀释,硅藻土过滤,二氯甲烷淋洗,向滤液中加入4 mol/mL HCl(260 mL)搅拌,调节pH至2~3,分层得水相。向水相中加入300 mL二氯甲烷搅拌,加入28%的NaOH溶液调节pH至10~11,分液,水相用300 mL二氯甲烷萃取,合并有机相,用饱和食盐水洗1次,Na2SO4干燥后浓缩得固体,丙酮打浆后抽滤得白色固体209.5 g,收率92.4%(HPLC含量99.2%)。熔点:198℃。1H NMR(400 MHz,CDCl3)δ 8.41(s,1H),8.28(s,1H),8.19(s,1H),7.79(d,J=12.3 Hz,1H),7.69(d,J=8.6,2.2 Hz,1H),7.26(s,1H),4.74(m,J=13.9,7.0 Hz,1H),3.50(s,3H),2.69(s,6H),2.49(m,4H),1.70(t,J=9.6 Hz,6H),1.08(t,J=7.2 Hz,3H)。
2 结果与讨论
2.1 异丙基乙酰胺(1)和(E)-N'-(4-溴-2,6-二氟苯基)-N-异丙基乙脒(2)的合成
氨基乙酰化试剂可以是乙酸酐和乙酰氯。但在异丙胺酰基化实验中发现,乙酸酐作为酰化试剂收率(93.8%)高于乙酰氯(80.0%),且没有HCl气体生成。在Et3N和POCl3存在下,4-溴-2,6-二氟苯胺作为亲核试剂进攻化合物1上的羰基,发生加成消除反应生成化合物2[6-7]。实验过程发现三氟乙酸可以替代三乙胺,促进反应的顺利进行。但三氟乙酸的价格较三乙胺贵,从经济角度考虑采用三乙胺。
2.2 6-溴-4-氟-1-异丙基-2-甲基-1H-苯并[d]咪唑(3)的合成
化合物2合成化合物3的反应机理如图2所示,在碱性条件下,化合物2中氨基上的H原子被拔去,形成负离子,经过对苯环上氟原子的亲核取代生成化合物3。

图2 化合物2生成3的反应机理Fig.2 Formation mechanism of compound 3 from 2
从机理来看,在化合物3的生成过程中,碱还可以作为亲核试剂,直接取代化合物2中的卤原子,生成副产物。因此,本研究主要考察了碱对反应的作用,同时也探讨了溶剂和温度对反应的影响,结果见表1。从表1中可以看出,以t-BuOK为碱,DMF为溶剂时,化合物2虽反应完全,但杂质居多,导致目标产物化合物3收率低(60.7%);而以THF为溶剂时,化合物2则完全转化为副产物。原因是t-BuOK的碱性太强,低温(THF沸点66℃)时,直接将化合物2苯环上的卤原子取代,而高温(100℃)有利于氨基的拔氢,生成化合物3。同时,以含有2%水的THF或DMF做溶剂时,反应几乎停止。说明水可以将t-BuOK转化为KOH,降低了体系的碱性,从而极大影响反应。
在合成苯并咪唑类化合物时,使用K2CO3、KOH、t-BuOK可以提供反应所需碱性环境[8-11]。但从本研究的结果来看,使用K2CO3作为碱时,无论选用哪种溶剂,基本没有反应。而使用KOH做为碱时,原料没有反应完全,收率最高只有35.6%;以THF为溶剂时,没有反应,说明弱碱既不能直接取代苯环上的卤素,也不能拔掉氨基的氢。同时,K2CO3和KOH在有机溶剂中的溶解性差,降低了其碱性。
t-BuONa碱性介于KOH和t-BuOK之间,结果显示以THF为反应溶剂时,反应进展缓慢,且全部转化为杂质;而以DMF为反应溶剂,温度为100℃时,4 h~5 h反应完全,产物收率达83.0%,相较于采用t-BuOK,收率大幅提高,反应时间大幅缩短。
2.3 4-氟-1-异丙基-2-甲基-6-(4,4,5,5-四甲基-1,3,2-二氧杂硼杂环戊烷-2-基)-1H-苯并[d]咪唑(4)和 6-(2-氯-5-氟嘧啶-4-基)-4-氟-1-异丙基-2-甲基-1H-苯并[d]咪唑(5)的合成
在催化剂 Pd(OAc)2存在下,3与双联硼酸频哪醇酯发生Miyaura硼烷基化反应生成4,利用Pd(PPh3)2Cl2为催化剂,4与 2,4-二氯-5-氟嘧啶发生 Suzuki偶合生成 5[10]。Pd(PPh3)2Cl2可催化卤代芳烃硼烷基化[12],本研究利用该催化剂也成功实现了3的硼烷基化。鉴于两步反应都可以Pd(PPh3)2Cl2作为催化剂,实验过程中研究了“一锅法”的可行性,即以 Pd(PPh3)2Cl2为催化剂,将合成4和5的两步反应原料一起投入反应瓶中进行反应,以期达到减少反应操作的目的。但是,实验发现化合物3完全转化为4和未知物,仅有少量5生成。笔者认为在化合物3、2,4-二氯-5-氟嘧啶和联硼酸频哪醇酯同时存在时,联硼酸频哪醇酯可以与3和4同时发生硼烷基化反应,不利于5的生成。
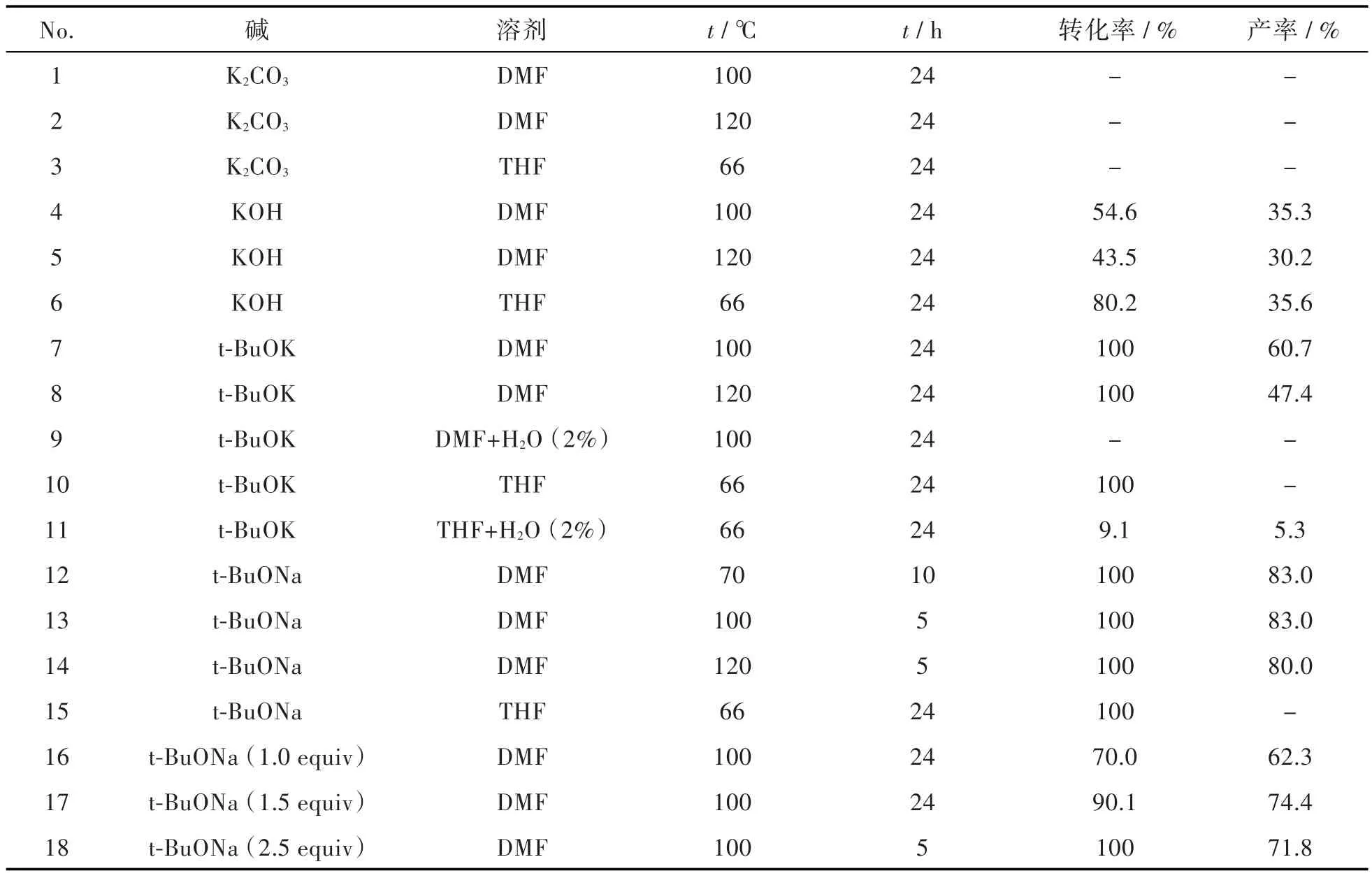
表1 不同反应条件对合成化合物3的影响aTab.1 Effects of reaction conditions on formation of compound 3a
2.4 1-((6-溴吡啶-3-基)甲基)-4-乙基哌嗪(6)的合成
6-溴-3-吡啶甲醛与N-乙基哌嗪发生还原胺化反应生成化合物 6。BH3·THF[13]和 NaBH(OAc)3[14]在还原胺化反应中有很好的效果,但是BH3·THF不适合用于放大生产。以NaBH(OAc)3为还原剂,实验结果表明反应不完全。加入钛酸异丙酯能够有效的促进反应[15],但在后处理过程中,钛酸异丙酯的除去过程导致收率特别低;加入催化量的乙酸[16],有助于催化反应。本研究,以 NaBH(OAc)3为还原剂,加入少量乙酸后,6-溴-3-吡啶甲醛可以完全转化为6,且分离操作简单。
2.5 (5-((4-乙基哌嗪-1-基)甲基)吡啶-2-胺(7)的合成
在氧化亚铜存在下,以液氨为氮源,或在2-(二环己基膦基)联苯(CyJohnPhos)、三(二亚苄基丙酮)二钯(Pd2(dba)3)的作用下,以六甲基二硅基氨基锂(LiHMDS)为氮源,化合物6吡啶环上的溴原子被氨基取代得到化合物7,前者收率为54.5%,后者收率为 49.1%[17-19]。以水为溶剂,在四丁基溴化铵、Cs2CO3和[Cu2(bpnp)(OH)(CF3COO)3](bpnp=2,7-双(吡啶-2-基)-1,8-二氮杂萘)存在的条件下,以液氨为氮源,2-溴吡啶100%转化为2-氨基吡啶[20]。以乙二醇为溶剂,N,N'-二甲基乙二胺为配体,在K2CO3和Cu2O的存在下,以氨水为氮源,3-溴吡啶 80%转化为 3-氨基吡啶[21]。以 DMSO做溶剂,添加K2CO3或者Cs2CO3,在CuI的存在下,以氨水为氮源,碘苯99%转化为苯胺[22]。
为提高化合物6生成7的转化率,并考虑成本因素,对原反应体系进行了改进,在K2CO3和Cu2O存在下,以DMEDA为配体,以氨水为氮源,考察不同溶剂对反应的影响(见表2)。
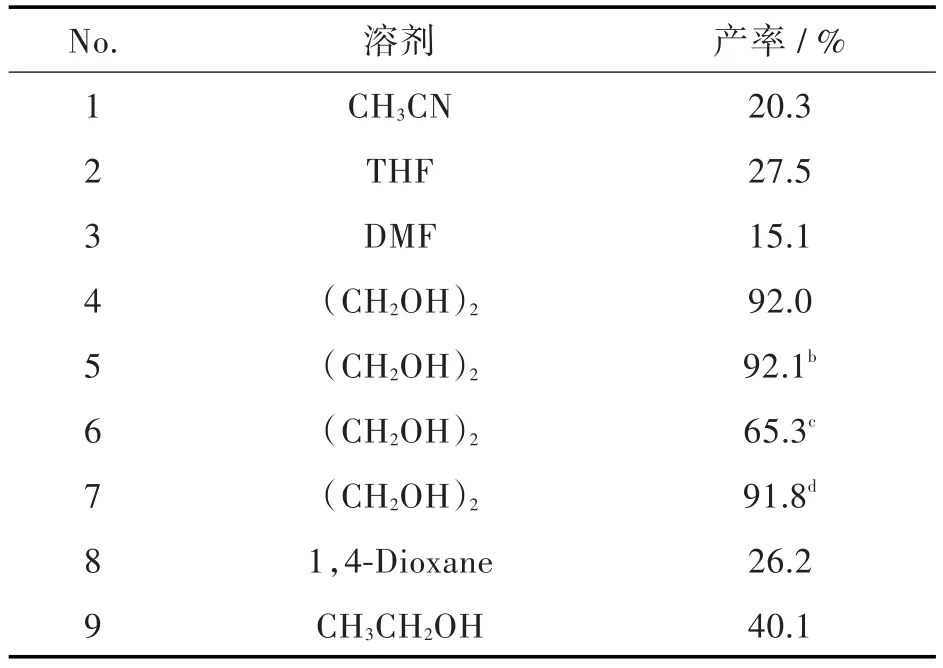
表2 不同溶剂对制备化合物7产率的影响aTab.2 The yields of compound 7 with different solventsa
结果表明,乙腈、四氢呋喃、乙醇、DMEDA作为溶剂时,反应进行的不完全;1,4-二氧六环和乙二醇作为反应溶剂时,反应都进行的彻底,但前者作溶剂时副产物多,而乙二醇作溶剂时收率可达92.0%。实验过程中发现乙二醇由于沸点高会残留在化合物7中,影响化合物7的纯度,但对后续反应没有影响。
2.6 Abemaciclib的合成
化合物5和7通过Buchwald-Hartwig反应生成abemaciclib,反应使用了 Cs2CO3提供碱性环境[5,18,23]。类似的偶联反应,也可以使用K2CO3提供碱性环境[17,19,24]。研究发现 K2CO3可以替代 Cs2CO3,促进 5和7偶联反应的顺利进行,而且Cs2CO3的价格较K2CO3贵,从经济角度考虑采用K2CO3。
3 结 语
本研究以4-溴-2,6-二氟苯胺和异丙胺为原料合成了中间体6-(2-氯-5-氟嘧啶-4-基)-4-氟-1-异丙基-2-甲基-1H-苯并[d]咪唑;以 N-乙基哌嗪和 6-溴-3-吡啶甲醛为原料反应合成了中间体(5-((4-乙基哌嗪-1-基)甲基)吡啶-2-胺。在前一个中间体的合成过程中,生成苯并咪唑时以t-BuONa为碱,产率可达83.0%;Miyaura硼烷基化和Suzuki偶联反应中,催化剂均采用PdCl2(PPh)2。在后一个中间体合成中,还原胺化反应中加入催化量的乙酸,原料反应更完全;吡啶环上溴原子的氨基取代反应中以乙二醇为溶剂,N,N'-二甲基乙二胺为配体,收率可达85.0%。两中间体经Buchwald-Hart wig偶联反应合成目标产物abemaciclib时,以K2CO3代替Cs2CO3为碱,收率可达92.4%。优化后的反应条件温和、操作时间短且产物易于纯化,提高了产率,降低了成本,适合工业化生产。
参考文献:
[1]DIGIULIO S. FDA's breakthrough therapy designation to abemaciclib for breast cancer[J].Oncology Times,2015,37(21):21.
[2]PATNAIKA,ROSEN L S,TOLANEY S M,et al.Abstract CT232:clinical activity of LY2835219,a novel cell cycle inhibitor selective for CDK4 and CDK6,in patients with metastatic breast cancer[J].Cancer Research,2014,74(19):CT232.
[3]KIM E S.Abemaciclib:First global approval[J].Drugs,2017,77(18):2063-2070.
[4]FREDERICK M O,KJELL D P. A synthesis of abemaciclib utilizing a Leuckart-Wallach reaction[J].Tetrahedron Letters,2015,56(7):949-951.
[5]张浩,张剑,陈奉泉.Abemaciclib合成路线图解[J].药物化学,2016,4(4):38-41.
[6]YUE X L,LI H,LIU S S,et al. N-fluorinated phenyl-N′-pyrimidylureaderivatives: synthesis,biological evaluation and 3D-QSAR study[J].Chinese Chemical Letters,2014,25(7):1069-1072.
[7]KATRITZKY A R,CAI C,SINGH S K. Efficient microwave access to polysubstitutedamidines from imidoylbenzotriazoles[J].Cheminform,2006,71(9):3375-3380.
[8]LIUBCHAK K,NAZARENKO K,TOLMACHEV A.Cheminform abstract: synthesis of annulated benzimidazoles via amidine cyclization[J].Tetrahedron,2012,68(14):2993-3000.
[9]INFANTE-CASYILLO R,HERNANDEA-RIVERA S P.Experimental and theoretical studies of the molecular structure of five new 2-methylbenzimidazole derivatives[M].New York:Nova Science Publishers,2011:11.
[10]蔡雄,钱长庚,刘斌,等.吡啶嘧啶胺类化合物或吡啶吡啶胺类化合物及其应用:中国,105153119A[P].2015-09-11.
[11]SANCHEZ-MARTINEZ C.Preparation of benzimidazo⁃lylpyrimidinylaminopyridines as CDK4/6 protein kinase inhibitors: US, 20100160340A1 [P].2010-07-24.
[12]DZHEVAKOV P B,TOPCHIY M A,ZHARKOVA D A,et al. Miyauraborylation and one-pot two-step homocoupling of aryl chlorides and bromides under solvent-freeconditions [J].Advanced Synthesis &Catalysis,2016,358(6):977-983.
[13]PLETZ J,BERG B,BREINBAUER R.Cheminform abstract:a general and direct reductive amination of aldehydes and ketones with electron-deficient anilines[J].Synthesis,2016,48(9):1301-1317.
[14]ABDELMAGID A F,CARSON K G,HARRIS B D,et al.Reductive amination of aldehydes and ketones with sodium triacetoxyborohydride.Studies on direct and indirectreductive amination procedures [J].Journal of Organic Chemistry,1996,61(11):38-49.
[15]MATTSON R J,PHAM K M,LEUCK D J,et al.Cheminform abstract:an improved method for reductive alkylation of amines using Titanium(IV)isopropoxide and sodium syanoborohydride [J].Cheminform,1990,21(41):2552-2554.
[16]BOSCHELLI D H,WU B,BARRISO S A C,et al.Inhibition ofsrc kinase activity by 7-[(2,4-dichloro-5-methoxyphenyl)amino]-2-heteroaryl-thieno[3,2-b]pyridine-6-carbonitriles[J].Bioorganic&MedicinalChemistry Letters, 2005, 15 (21) :4681-4687.
[17]COATES D A,GELBERT L M,KNOBELOCH J M,et al. Protein kinase inhibitors:US,7855211[P].2010-12-21.
[18]ANDREW C D,ALFONSO D,ANA D,et al. Protein kinase inhibitors:WO,2010075074A1[P].2010-07-24.
[19]CHAN E M. Combination therapy for cancer:WO,2015130540A1[P].2015-09-03.
[20]LIAO B S,LIU S T. Homogenous bimetallic catalysis on amination of ArX and ArX2in aqueous medium-synergistic effect of dicopper complexes[J].Catalysis Communications,2013,32(5):28-31.
[21]ELMKADDEM M K,FISCHMEISTER C,THOMAS C M,et al. Cheminform abstract:efficient synthesis ofaminopyridine derivativesby coppercatalyzed amination reactions[J].Chemical Communications,2010,46(6):925.
[22]WANG Y,LUO J,LIU Z .Cheminform abstract:synthesis of a novel 8-hydroxyquinoline functionalized poly(ethylene glycol) bridged dicationic ionic liquid and its application in palladium-catalyzed Heck reaction under solvent-free conditions[J].Journal of Organometallic Chemistry,2014,44(46):1-5.
[23]WOLINSKA E. Sequential amination of heteroaromatic halides with aminopyridine 1-oxides and theirN-protected derivativesbased on novel aza-smiles rearrangement[J].Heterocyclic Communications,2012,18(5/6):227-232.
[24]BONNATERRE F,BOIS-CHOUSSY M,ZHU J.Rapid access to oxindoles by the combined use of an ugifour-component reaction and a microwave-assisted intramolecular Buchwald-Hartwig amidation reaction[J].Organic Letters,2006,8(19):4351-4354.
猜你喜欢
磷肥与复肥(2022年5期)2022-06-18
能源化工(2021年3期)2021-12-31
化工设计通讯(2020年7期)2020-07-25
山东化工(2020年9期)2020-06-01
天然产物研究与开发(2018年8期)2018-09-10
山东工业技术(2016年16期)2016-08-22
中国资源综合利用(2016年7期)2016-02-03
中国塑料(2015年12期)2015-10-16
火炸药学报(2015年6期)2015-03-08
有机氟工业(2015年3期)2015-03-03
