
Complete genome of Cobetia marina JCM 21022 T and phylogenomic analysis of the family Halomonadaceae*
2018-05-07 06:07TANGXianghai唐祥海XUKuipeng徐奎鹏HANXiaojuan韩晓娟MOZhaolan莫照兰MAOYunxiang茅云翔
TANG Xianghai (唐祥海) XU Kuipeng (徐奎鹏) HAN Xiaojuan (韩晓娟) MO Zhaolan (莫照兰) MAO Yunxiang (茅云翔)
1 Ministry of Education Key Laboratory of Marine Genetics and Breeding, College of Marine Life Sciences, Ocean University of China, Qingdao 266003, China
2 Key Laboratory of Sustainable Development of Marine Fisheries, Ministry of Agriculture, Yellow Sea Fisheries Research Institute, Chinese Academy of Fishery Sciences, Qingdao 266071, China
3 CAS Key Laboratory of Biobased Materials, Qingdao Institute of Bioenergy and Bioprocess Technology, Chinese Academy of Sciences, Qingdao 266101, China
1 INTRODUCTION
Cobetiamarinais a gram-negative marine bacterium that is used as a model organism in marine biofouling studies for its typical obligatory aerobic characteristics that allows easy culturing and handling.Further, biofilms formed byC.marinacan influence the secondary colonization of invertebrates and algae(Shea et al., 1995). The chronological record of the taxonomy shows thatC.marinahas hitherto been classified and revised many times. Although all the classifications referred to the same species, it was first identified asArthrobactermarinus(Cobet et al.,1970), and subsequently described asPseudomonasmarina(Baumann et al., 1972),Delayamarina(Baumann et al., 1983; Shea et al., 1995; Ista et al.,1996) andHalomonasmarina(Dobson and Franzmann, 1996; Ista et al., 1999; Mata et al., 2002).More than ten years ago, it was suggested thatHalomonasmarinashould be assigned to a new genusCobetiain the family Halomonadaceae based on a comparative sequence analysis of 23S and 16S rRNA(Arahal et al., 2002a, b) sequences. This was because theHalomonasmarinasequences were too phylogenetically distant from those of otherHalomonasspecies to be considered as belonging to the same genus.
To date, although many molecular studies have been carried out to understand the phylogenetic relationship of Halomonadaceae species, these studies were based mainly on 23S and 16S rDNA sequences(Dobson and Franzmann, 1996; Arahal et al., 2002a,b; Ntougias et al., 2007; de la Haba et al., 2010;Romanenko et al., 2013). Therefore, to better understand theC.marinasurface-associated lifestyle and the phylogeny of the family Halomonadaceae, it is important to sequence the whole genomes of representative species and strains. The genome sequences of several Halomonadaceae species have been reported recently (Copeland et al., 2011;Schwibbert et al., 2011; Sánchez-Porro et al., 2013;Sharko et al., 2016), including the genome ofC.marinaKMM 296 (Balabanova et al., 2016a) that was afterward reclassified asC.amphilectiKMM 296(Balabanova et al., 2016b). In this study, we used the publicly available genome information to conduct comparative genomics and phylogenomics analyses.On strategy of genome sequencing and assembly, the single molecule real-time sequencing technology(SMRT) was chosen in this study, which is a sequencing-by-synthesis technology based on realtime imaging of fluorescently tagged nucleotides as they are synthesized along individual DNA template molecules. SMRT can produces long and unbiased sequences, and ensure assembly of complex repeat structures and GC and AT rich regions that are often hard to assemble in short-read sequencing technology(Eid et al., 2009; Roberts et al., 2013).
In this study, the de novo assembly and annotation of the complete genome sequence ofC.marinastrain JCM 21022Tsuggested this strain had a circular chromosome that contained a set of crucial genes involved in processes related to surface attachment.In addition, the comparative genomics and phylogenomics analyses revealed genome structural features and allowed us to reconstruct the phylogenetic relationships of species in the family Halomonadaceae.
2 MATERIAL AND METHOD
2.1 DNA extraction, sequencing, and genome assembly
TheC.marinastrain JCM 21022T(=DSM 4741T=ATCC 25374T) was obtained from RIKEN Bio Resource Center (BRC) and grown on Marine agar 2216 (BD-Difco). Genomic DNA was extracted using the SDS method combined with RNase A treatment(Wilson, 1997).
Genomic DNA was fragmented, end-repaired, and SMRT bell DNA template libraries (insert size about 10 Kb) were prepared according to the manufacturer’s specifications. SMRT sequencing (two SMRT cells)was performed on the Pacific Biosciences RS II sequencer using the P4-C2 chemistry according to standard protocols.
Pacbio long reads were obtained from the two SMRT sequencing cells. Reads longer than 1 000 bp with quality values greater than 0.8 were merged.Then the filtered reads were analyzed using the hierarchical genome assembly process (HGAP3)pipeline (Chin et al., 2013). The pipeline uses the longest reads as seeds to recruit all other reads for the construction of highly accurate preassembled reads that were then assembled using the Celera Assembler.To reduce the numbers of remaining insertions or deletions (indels) and base substitution errors in the draft assembly, the Quiver consensus algorithm was used to derive a highly accurate consensus for the final assembly.
2.2 Genome annotation
The location of the replication origins was predicted using the Ori-Finder web service (Gao and Zhang,2008). Open reading frame (ORF) prediction and annotation were performed using MicroScope, the microbial genome annotation and analysis platform(Vallenet et al., 2013), and verified using Glimmer(Salzberg et al., 1998), RNAmmer (Lagesen et al.,2007) and tRNAscan-SE (Lowe and Eddy, 1997). A graphical circular map of the genome was drawn with Circos v0.66 (Krzywinski et al., 2009).
2.3 Comparative genome analysis
We compared the genome sequences ofC.marinaJCM 21022TandC.amphilectiKMM 296(Balabanova et al., 2016a) with BLAST searches(E-value 1.0e-5) to identify similarity at the nucleotide level (Camacho et al., 2009) and with MUMmer 3.23 for a detailed collinearity analysis at the amino acid level (Kurtz et al., 2004). The comparative study on the pan and core genome was performed using BLASTP and SiLiX software(Camacho et al., 2009; Miele et al., 2011).

Table 1 Bacterial genome sequences used in this study
2.4 Phylogenetic analyses
To elucidate the phylogenetic relationship ofC.marinaJCM 21022Tamong species in Halomonadaceae, the concentrated amino acid sequences from 13 species were used to construct a phylogenetic tree. The sequences included 12 complete genomes from Halomonadaceae. TheEscherichiacoligenome was used to root the tree (Table 1). The sequences were aligned using MAFFT version 5 and adjusted manually (Katoh et al., 2005). All the aligned sites were trimmed using trimAl v1.4 with the options“-automated1” (Capella-Gutiérrez et al., 2009).RAxML61 was employed to reconstruct a maximum likelihood phylogenetic tree for each cluster with an evolutionary model specified as ‘PROTGAMMALG’according to PROTTEST-3.4, and a bootstrap significance test was performed with 1 000 replicates(Stamatakis, 2006; Darriba et al., 2011).
3 RESULT
3.1 Genome assembly and annotation
SMRT sequencing ofC.marineJCM 21022Tgenerated 224 627 high quality long reads(1 861 380 354 bp) with a coverage of 465×. The HGAP3 pipeline was used to correct sequencing errors. The resulting corrected reads were assembled into a single 4 176 300 bp long contig with an average GC content of 62.44% and 4.07% repeat regions(Fig.1). TheC.marinagenome sequence was annotated automatically using the MicroScopeplatform. A total of 3 611 predicted protein-coding sequences (CDSs) with an average length of 1 002 bp were identified as well as 21 rRNA operons (16S,23S, and 5S) and 72 predicted tRNAs genes (Table 2).About 33.70% of the open reading frames (ORFs)were predicted to code hypothetical proteins (1 217 ORFs). No plasmids were identified and no clustered regularly interspaced short palindromic repeat(CRISPR) loci were detected in the genome.

Table 2 Genomic characteristics of the marine bacterium Cobetia marina JCM 21022 T
The predicted CDSs were annotated with the cluster of orthologous groups (COG) database (Fig.2).The main annotations were “Amino acid transport and metabolism” (404 genes), “General function prediction only” (471 genes), “Transcription” (260 genes), “Carbohydrate transport and metabolism”(242 genes), “Cell wall/membrane/envelope biogenesis” (220 genes), “Inorganic ion transport and metabolism” (221 genes), and “Translation, ribosomal structure and biogenesis” (183 genes). Further analysis revealed many important genes in theC.marinagenome that were related to its ecophysiological status; for example, genes related to cell-surface structures that encoded proteins for curli(Barnhart and Chapman, 2006) and Type IV pili(Spangenberg et al., 1995), and a gene that encoded O-antigen polymerase for extracellular polymer components (Wang and Reeves, 1998). These genes are known to be important for surface attachment in other organisms. In addition, several genes involved in the stress response were also identified in theC.marinagenome, including genes coding RpoS,universal stress protein A, stringent starvation proteins(Thomas et al., 2008).
3.2 Comparative genome analysis
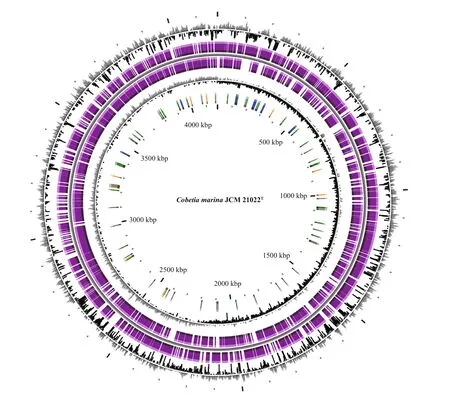
Fig.1 Graphical circular map of the Cobetia marina JCM 21022 T chromosome
We compared theC.marinaJCM 21022Tgenome with theC.amphilectiKMM 296 genome. The results show only about 67% similarity at the nucleotide level across the two genomes (Fig.3). The differences were mainly a result of indels in the sequences. The alignment of the two genomes at the amino acid level revealed several chromosomal recombination or transposition and inversions (Fig.4). A comparative study on the pan and core genes was conducted by comparing the genome sequences of the two strains.As a result, 5 931 core genes (2 646 families) and 7 482 pan genes (4 169 families) were obtained,respectively (Table 3).C.marinaJCM 21022Tshared 2 962 of 3 611 (82.03%) homologous genes withC.amphilectiKMM 296, implying similar gene functions between them. We detected 645 and 906 strain-specific genes inC.marinaJCM 21022TandC.amphilectiKMM 296 respectively.
3.3 Phylogenetic relationship
To elucidate the evolutionary position ofC.marinaJCM 21022Tin family Halomonadaceae, a data set of 635 concentrated single-copy genes were selected based on the alignment of the genome sequences of 13 related species and its corresponding amino acid sequences were used to construct the phylogenetic tree. The maximum likelihood tree (ML) recovered a well-supported classification among different clades(Fig.5). The phylogenomic study showed thatC.marinaJCM 21022Tclustered withC.amphilectiKMM 296, indicating a close relationship. The polygenetic tree also showed that species of genusHalomonasclustered together withChromohalobacter,and this cluster then emerged as a sister group to the genusCobetia. Additionally, the clade unitingHalotaleaalkalilentaIHBB 13600 andZymobacter palmaeDSM 10491Twas strongly supported by the bootstrap results, and emerged as a sister group toKushneriaaurantiaDSM 21353Twith high confidence. This group diverged from the other nine species of Halomonadaceae at the base of the phylogenetic tree.
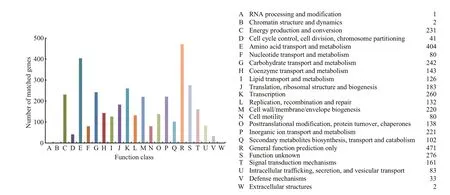
Fig.2 Clusters of orthologous group annotations for the Cobetia marina JCM 21022 T genome

Fig.3 Comparison of the Cobetia marina JCM 21022 T and Cobetia amphilecti KMM 296 genomes at the nucleotide level by BLAST search

Fig.4 Collinearity study of the Cobetia marina JCM 21022 T and Cobetia amphilecti KMM 296 genomes at the amino acid level

Fig.5 Maximum likelihood phylogenetic tree for Cobetia marina JCM 21022 T and the genomes of 12 related species
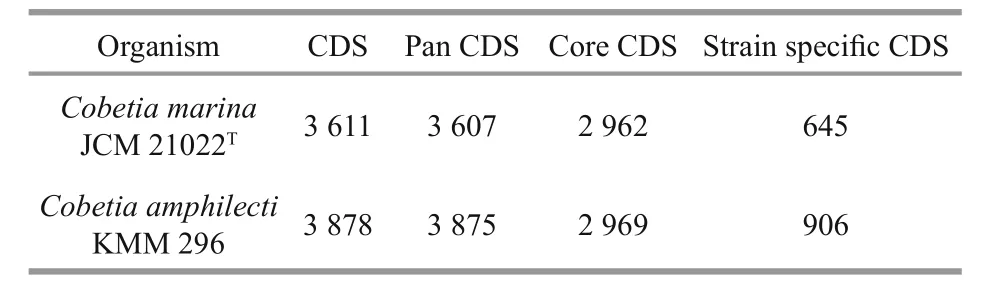
Table 3 Pan- and core-genes in the Cobetia marina JCM 21022 T and Cobetia amphilecti KMM 296 genomes
4 DISCUSSION
In this study, we first presented the whole genomic characteristics ofC.marinaJCM 21022T, and annotated the predicted genes to several metabolismrelated COG categories. BecauseC.marinais used as a model organism in marine biofouling studies, its successful attachment on the host surface must be the primary step in biofilm formation (Shea et al., 1995;Arpa Sancet, 2013). As expected, genes that encode for cell-surface structures and extracellular polymer components were detected in theC.marinaJCM 21022Tgenome. The presence of genes that encode curli proteins suggests thatC.marinaJCM 21022Tcan attach to invertebrates and algae host surfaces by producing curli, which has been indicated to play significant roles in the interactions betweenE.coliorSalmonellawith plant surfaces (Barak et al., 2005;Jeter and Matthysse, 2005).
In addition, we compared theC.marinaJCM 21022TandC.amphilectiKMM 296 (formerly namedC.marinaKMM 296) genome sequences. Recently,clusters of genes were detected in theC. amphilectiKMM 296 genome that are involved in the transport and metabolism of nitrogen, sulfur, iron, and phosphorus compounds, which have been shown to provide alternative capacity of the inhabitants of the rhizospheres of terrestrial plants, as well as deep-sea ecological communities (Balabanova et al., 2016a).As expected, these genes were also observed in theC.marinaJCM 21022Tgenome. Despite the numerous common genes described above, we identified multiple chromosomal recombination or transposition and inversions between the two genomes. Considering these observations and the relatively large numbers of strain-specific genes, we inferred that there are differences in physiological characteristics and ecological adaptations amongC.marinastrains,which is consistent with previous studies (Ivanova et al., 2005). Therefore, we consider that it is reasonable to revise the classification ofC.marinaKMM 296 asC.amphilectiKMM 296 (Balabanova et al., 2016b).
Considering whole genomes can provide more sequence signatures for use in phylogenetic analysis than restricted rDNA sequence, we used 13 bacterial genomes to construct the polygenetic tree of Halomonadaceae. Our results indicated that the genusHalomonas(Vreeland et al., 1980) clustered withChromohalobacter(Ventosa et al., 1989) first, and then withCobetia, which was clearly distinguished from the former two groups. Moreover, the phylogenetic relations of other genera such asKushneria(Sánchez-Porro et al., 2009),Zymobacter(Okamoto et al., 1993), andHalotalea(Ntougias et al., 2007, 2015) were also well resolved. Therefore,we have shown that whole genome information is an appropriate tool in the phylogenetic analysis of bacteria for its ability to provide more evolutionary information than rDNA sequences alone.
In summary, in this study, we sequenced,assembled, and annotated the complete genome sequence ofC.marinastrain JCM 21022T, and detected a set of crucial genes that may be involved in surface attachment related processes. Although similar gene functions were revealed betweenC.marinaJCM 21022TandC.amphilectiKMM 296(formerly namedC.marinaKMM 296), the significant differences identified in the comparative genome analysis resulted from sequence indels and chromosomal recombination. In addition, the phylogenomic study of species in the family Halomonadaceae based on whole genome information was conducted here for the first time,and the relationships among every genus were well resolved. In future studies to comprehensively understand the surface-associated lifestyle ofC.marinaJCM21022T, genes or gene clusters that endow this species important physiological features should be identified.
Arahal D R, Castillo A M, Ludwig W et al. 2002a. Proposal ofCobetiamarinagen. nov., comb. nov., within the family Halomonadaceae, to include the speciesHalomonas marina.SystematicandAppliedMicrobiology,25(2):207-211.
Arahal D R, Ludwig W, Schleifer K H et al. 2002b. Phylogeny of the family Halomonadaceae based on 23S and 165 rDNA sequence analyses.InternationalJournalof SystematicandEvolutionaryMicrobiology,52(1): 241-249.
Arpa Sancet M P. 2013. Influence of surface properties on adhesion ofCobetiamarinaand accumulation of marine microfoulers in the ocean. Ruperto Carola University Heidelberg, Heidelberg.
Balabanova L A, Golotin V A, Kovalchuk S N et al. 2016a. The Genome of the marine bacteriumCobetiamarinaKMM 296 isolated from the musselCrenomytilusgrayanus(Dunker, 1853).RussianJournalofMarineBiology,42(1): 106-109.
Balabanova L, Nedashkovskaya O, Podvolotskaya A et al.2016b. Data supporting functional diversity of the marine bacteriumCobetiaamphilectiKMM 296.DatainBrief,8: 726-732.
Barak J D, Gorski L, Naraghi-Arani P et al. 2005.Salmonella entericavirulence genes are required for bacterial attachment to plant tissue.AppliedandEnvironmental Microbiology,71(10): 5 685-5 691.
Barnhart M M, Chapman M R. 2006. Curli biogenesis and function.AnnualReviewofMicrobiology,60: 131-147.
Baumann L, Baumann P, Mandel M et al. 1972. Taxonomy of aerobic marine eubacteria.JournalofBacteriology,110(1): 402-429.
Baumann L, Bowditch R D, Baumann P. 1983. Description ofDeleyagen. nov. created to accommodate the marine speciesAlcaligenesaestus,A.pacificus,A.cupidus,A.venustus, andPseudomonasmarina.International JournalofSystematicandEvolutionaryMicrobiology,33(4): 793-802.
Camacho C, Coulouris G, Avagyan V et al. 2009. BLAST+:architecture and applications.BMCBioinformatics,10:421.
Capella-Gutiérrez S, Silla-Martínez J M, Gabaldón T. 2009.trimAl: a tool for automated alignment trimming in largescale phylogenetic analyses.Bioinformatics,25(15):1 972-1 973.
Chin C S, Alexander D H, Marks P et al. 2013. Nonhybrid,finished microbial genome assemblies from long-read SMRT sequencing data.NatureMethods,10(6): 563-569.
Cobet A B, Wirsen C Jr, Jones G E. 1970. The effect of nickel on a marine bacterium,Arthrobactermarinussp. nov.JournalofGeneralMicrobiology,62(2): 159-169.
Copeland A, O’Connor K, Lucas S et al. 2011. Complete genome sequence of the halophilic and highly halotolerantChromohalobactersalexigenstype strain (1H11T).StandardsinGenomicSciences,5(3): 379-388.
Darriba D, Taboada G L, Doallo R et al. 2011. ProtTest 3: fast selection of best-fit models of protein evolution.Bioinformatics,27(8): 1 164-1 165.
de la Haba R R, Arahal D R, Márquez M C et al. 2010.Phylogenetic relationships within the family Halomonadaceae based on comparative 23S and 16S rRNA gene sequence analysis.InternationalJournalof SystematicandEvolutionaryMicrobiology,60(4): 737-748.
Dobson S J, Franzmann P D. 1996. Unification of the generaDeleya(Baumann et al. 1983),Halomonas(Vreeland et al. 1980), andHalovibrio(Fendrich 1988) and the speciesParacoccushalodenitrificans(Robinson and Gibbons 1952) into a single genus,Halomonas, and placement of the genusZymobacterin the familyHalomonadaceae.InternationalJournalofSystematicBacteriology,46(2):550-558.
Eid J, Fehr A, Gray J et al. 2009. Real-time DNA sequencing from single polymerase molecules.Science,323(5910):133-138.
Gao F, Zhang C T. 2008. Ori-Finder: a web-based system for findingoriCs in unannotated bacterial genomes.BMC Bioinformatics,9: 79.
Ista L K, Fan H Y, Baca O et al. 1996. Attachment of bacteria to model solid surfaces: oligo(ethylene glycol) surfaces inhibit bacterial attachment.FEMSMicrobiologyLetters,142(1): 59-63.
Ista L K, Pérez-Luna V H, López G P. 1999. Surface-grafted,environmentally sensitive polymers for biofilm release.AppliedandEnvironmentalMicrobiology,65(4): 1 603-1 609.
Ivanova E P, Christen R, Sawabe T et al. 2005. Presence of ecophysiologically diverse populations withinCobetia marinastrains isolated from marine invertebrate, algae and the environments.MicrobesandEnvironments,20(4):200-207.
Jeter C, Matthysse A G. 2005. Characterization of the binding of diarrheagenic strains ofE.colito plant surfaces and the role of curli in the interaction of the bacteria with alfalfa sprouts.MolecularPlant-MicrobeInteractions,18(11):1 235-1 242.
Katoh K, Kuma K I, Toh H et al. 2005. MAFFT version 5:improvement in accuracy of multiple sequence alignment.NucleicAcidsResearch,33(2): 511-518.
Krzywinski M, Schein J, Birol I et al. 2009. Circos: an information aesthetic for comparative genomics.Genome Research,19(9): 1 639-1 645.
Kurtz S, Phillippy A, Delcher A L et al. 2004. Versatile and open software for comparing large genomes.Genome Biology,5(2): R12.
Lagesen K, Hallin P, Rødland E A et al. 2007. RNAmmer:consistent and rapid annotation of ribosomal RNA genes.NucleicAcidsResearch,35(9): 3 100-3 108.
Lowe T M, Eddy S R. 1997. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence.NucleicAcidsResearch,25(5): 955-964.
Mata J A, Martínez-Cánovas J, Quesada E et al. 2002. A detailed phenotypic characterisation of the type strains ofHalomonasspecies.SystematicandAppliedMicrobiology,25(3): 360-375.
Miele V, Penel S, Duret L. 2011. Ultra-fast sequence clustering from similarity networks with SiLiX.BMCBioinformatics,12: 116.
Ntougias S, Lapidus A, Copeland A et al. 2015. High-quality permanent draft genome sequence of the extremely osmotolerant diphenol degrading bacteriumHalotalea alkalilentaAW-7T, and emended description of the genusHalotalea.StandardsinGenomicSciences,10: 52.
Ntougias S, Zervakis G I, Fasseas C. 2007.Halotalea alkalilentagen. nov., sp. nov., a novel osmotolerant and alkalitolerant bacterium from alkaline olive mill wastes,and emended description of the familyHalomonadaceaeFranzmannetal. 1989, emend. Dobson and Franzmann 1996.InternationalJournalofSystematicand EvolutionaryMicrobiology,57(9): 1 975-1 983.
Okamoto T, Taguchi H, Nakamura K et al. 1993.Zymobacter palmaegen. nov., sp. nov., a new ethanol-fermenting peritrichous bacterium isolated from palm sap.Archives ofMicrobiology,160(5): 333-337.
Roberts R J, Carneiro M O, Schatz M C. 2013. The advantages of SMRT sequencing.Genomebiology,14: 405.
Romanenko L A, Tanaka N, Svetashev V I et al. 2013.Description ofCobetiaamphilectisp. nov.,Cobetia litoralissp. nov. andCobetiapacificasp. nov.,classification ofHalomonashaloduransas a later heterotypic synonym ofCobetiamarinaand emended descriptions of the genusCobetiaandCobetiamarina.InternationalJournalofSystematicandEvolutionary Microbiology,63(1): 288-297.
Salzberg S L, Delcher A L, Kasif S et al. 1998. Microbial gene identification using interpolated Markov models.Nucleic AcidsResearch,26(2): 544-548.
Sánchez-Porro C, de la Haba R R, Cruz-Hernández N et al.2013. Draft Genome of the marine GammaproteobacteriumHalomonastitanicae.GenomeAnnouncements,1(2):e00083-13.
Sánchez-Porro C, de la Haba R R, Soto-Ramírez N et al. 2009.Description ofKushneriaaurantiagen. nov., sp. nov., a novel member of the family Halomonadaceae, and a proposal for reclassification ofHalomonasmarisflaviasKushneriamarisflavicomb. nov., ofHalomonas indalininaasKushneriaindalininacomb. nov. and ofHalomonasavicenniaeasKushneriaavicenniaecomb.nov.InternationalJournalofSystematicandEvolutionary Microbiology,59(2): 397-405.
Schwibbert K, Marin-Sanguino A, Bagyan I et al. 2011. A blueprint of ectoine metabolism from the genome of the industrial producerHalomonaselongataDSM 2581T.EnvironmentalMicrobiology,13(8): 1 973-1 994.
Sharko F S, Shapovalova A A, Tsygankova S V et al. 2016.Draft genome sequence of “Halomonaschromatireducens”Strain AGD 8-3, a Haloalkaliphilic Chromate- and Selenite-Reducing Gammaproteobacterium.Genome Announcements,4(2): e00160-16.
Shea C, Lovelace L J, Smith-Somerville H E. 1995.Deleya marinaas a model organism for studies of bacterial colonization and biofilm formation.JournalofIndustrial Microbiology,15(4): 290-296.
Spangenberg C, Fislage R, Sierralta W et al. 1995. Comparison of type IV-pilin genes ofPseudomonasaeruginosaof various habitats has uncovered a novel unusual sequence.FEMSMicrobiologyLetters,125(2-3): 265-273.
Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihoodbased phylogenetic analyses with thousands of taxa and mixed models.Bioinformatics,22(21): 2 688-2 690.
Thomas T, Evans F F, Schleheck D et al. 2008. Analysis of thePseudoalteromonastunicatagenome reveals properties of a surface-associated life style in the marine environment.PLoSOne,3(9): e3252.
Vallenet D, Belda E, Calteau A et al. 2013. MicroScope—an integrated microbial resource for the curation and comparative analysis of genomic and metabolic data.NucleicAcidsResearch,41(D1): D636-D647.
Ventosa A, Gutierrez M C, Garcia M T et al. 1989. Classification of ‘Chromobacteriummarismortui’inanewgenus,Chromohalobactergen. nov., asChromohalobacter marismortuicomb. nov., nom. rev.InternationalJournal ofSystematicBacteriology,39(4): 382-386.
Vreeland R H, Litchfield C D, Martin E L et al. 1980.Halomonaselongata, a new genus and species of extremely salt-tolerant bacteria.InternationalJournalof SystematicBacteriology,30(2): 485-495.
Wang L, Reeves P R. 1998. Organization ofEscherichiacoliO157 O antigen gene cluster and identification of its specific genes.InfectionandImmunity,66(8): 3 545-3 551.
Wilson K. 1997. Preparation of genomic DNA from bacteria.In: Ausubel F M, Bent R, Kingston R E et al eds. Current Protocols in Molecular Biology. John Wiley & Sons, Inc.,New York. p.2.4.1-2.4.5.
 Journal of Oceanology and Limnology2018年2期
Journal of Oceanology and Limnology2018年2期
- Journal of Oceanology and Limnology的其它文章
- An aftereffect of global warming on tropical Pacific decadal variability*
- Analysis of monthly variability of thermocline in the South China Sea*
- A numerical study of the South China Sea Warm Current during winter monsoon relaxation*
- Predicting the sinkage of a moving tracked mining vehicle using a new rheological formulation for soft deep-sea sediment*
- Chemical characterization of fractions of dissolved humic substances from a marginal sea—a case from the Southern Yellow Sea*
- The morphological and molecular detection for the presence of toxic Cylindrospermopsis (Nostocales, Cyanobacteria) in Beijing city, China*