
山东省医疗器械生产企业《医疗器械临床试验质量管理规范》实施现状调研
2018-08-23 01:07陈洪忠张云娟山东省食品药品监督管理局审评认证中心器械检查科山东济南250000
中国医疗器械信息 2018年13期
陈洪忠 张云娟 山东省食品药品监督管理局审评认证中心 器械检查科 (山东 济南 250000)
内容提要: 目的:全面掌握我省医疗器械生产企业医疗器械临床试验开展现状,《医疗器械临床试验质量管理规范》实施现状,提高省内企业医疗器械临床试验的质量。方法:针对山东省的医疗器械生产企业设计专门的调研问卷,开展问卷调查,用Excel软件进行调研数据的统计处理。结果与结论:省内医疗器械生产企业基本能够执行《医疗器械临床试验质量管理规范》,规范实施后更加注重临床试验检查员队伍建设、临床试验团队更加壮大、专业化程度提高,更加重视对医疗器械临床试验相关法律法规的培训,对临床监查员专业要求更加严格。
为加强对医疗器械临床试验的管理,维护医疗器械临床试验过程中受试者权益,保证医疗器械临床试验过程规范,结果真实、科学、可靠和可追溯,根据《医疗器械监督管理条例》,国家食品药品监督管理总局制定《医疗器械临床试验质量管理规范》(以下简称《规范》),自2016年6月1日起施行。要求在中华人民共和国境内开展医疗器械临床试验,应当遵循本《规范》[1]。
为贯彻落实《国务院关于改革药品医疗器械审评审批制度的意见》(国发[2015]44号)要求,加强医疗器械临床试验监督管理,促进临床试验相关法律法规的实施,提高医疗器械临床检查质量、效率,提升检查尺度一致性,全面掌握我省医疗器械临床试验检查情况,山东省食品药品监督管理局审评认证中心结合实际工作,针对山东省内开展临床试验的医疗器械生产企业开展了一系列调研工作。本次调研问卷由各市局转发至各生产企业,并由各市局收集后转交我中心或者由企业直接传真至我中心。本次调研数据涵盖省内大中小各类医疗器械生产企业,数据具有广泛的代表性。本次调研内容包括企业对《医疗器械临床试验质量管理规范》的执行情况,《医疗器械临床试验质量管理规范》实施前后企业临床试验团队建设情况、项目实施情况、参加培训情况等多个项目。调研情况如下。
1.《规范》实施后对申办方医疗器械临床试验的影响
1.1 《规范》实施后医疗器械临床试验相关人员与申办方的配合程度
调研发现,随着《规范》的实施以及国家局和省局对临床试验重视程度的不断提升,临床试验机构及研究者面临着越来越严格的监管和问责,有70%生产企业认为选择临床试验机构变难,60%的生产企业认为研究者的配合程度下降,50%的生产企业认为临床试验机构的配合程度下降,66%的生产企业认为伦理委员会的配合程度下降。
1.2 《规范》实施后省内企业医疗器械临床试验的数量
《规范》的实施及国家局“医疗器械免临床目录”公布,约有30%的申办方表示其医疗器械临床试验的实施受到影响,实际进行临床试验数量比计划进行的临床试验数量减少,其中二类临床试验数量平均减少约40%,三类临床试验平均减少约20%,其中一家取消了全部计划的临床试验。
1.3 《规范》实施后申办方对医疗器械临床试验的重视程度
由以上数据可以看出,《医疗器械临床试验质量管理规范》的实施以及国家局和省局对医疗器械临床试验重视,直接引导了申办方更加重视医疗器械临床试验。具体表现如下。
1.3.1 申办方更加注重临床试验队伍建设
①申办方的临床试验团队更加壮大。总人数由规范实施后比规范实施前增长了20%;其中专职人员增长了21%;兼职人员18%。
②申办方的临床试验团队人员的专业化程度升高,如药学专业人员增长26%、临床医学专业人员增长15%、生物医学工程专业人员增长25%、统计学专业人员增长30%等。见图1。
③申办方的临床试验团队人员的整体受教育程度提高,目前本科以上学历已经占到临床试验团队人员的70%,人员的知识结构已处于较高水平。新规范实施后,本科学历人增长了15%,硕士学历人员由36%。
④申办方按规范要求,对临床试验监察员(CRA)专业要求更加严格,企业对有CRA专业进行要求的比例由原来的52%增长到77%,提升了15%。
1.3.2 申办方更加重视医疗器械临床试验相关制度建设
①申办方对医疗器械临床试验的标准操作更加规范。
规范实施前建立《医疗器械临床试验标准操作规程》的企业只有24家,仅占48%,而规范实施后建立《医疗器械临床试验标准操作规程》的企业占比达到了78%。见图2。
②申办方建立医疗器械临床试验监查标准操作规程的数量提高。
申办方建立医疗器械临床试验监查标准操作规程的数量提高了58%,目前已经有超过70%的申办方建立了自己的医疗器械临床试验监查标准操作规程。
1.3.3 申办方更加重视对医疗器械临床试验相关法律法规的培训
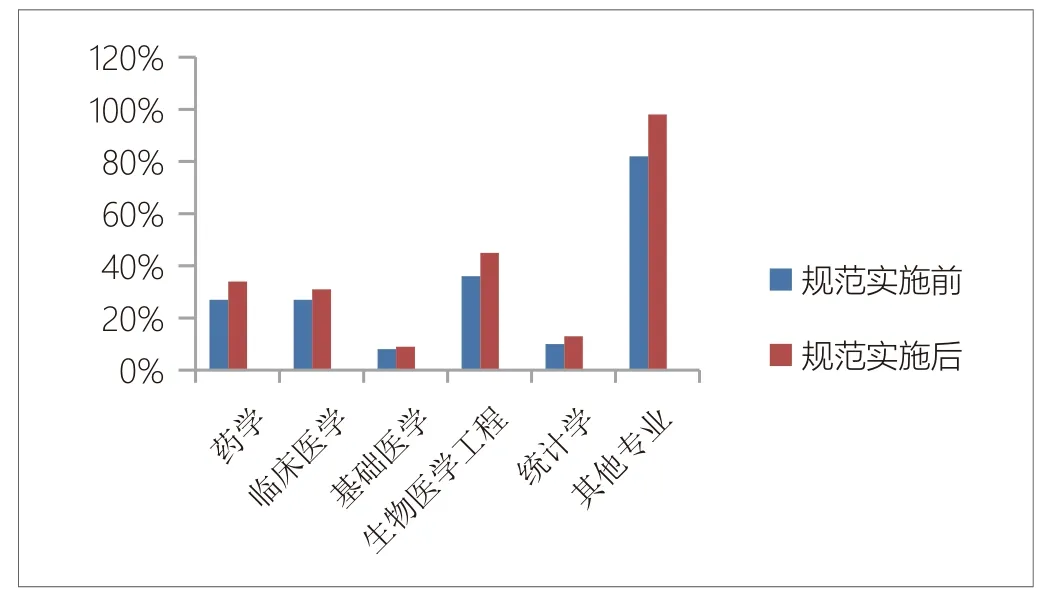
图1. 规范实施前后医疗器械生产企业临床试验人员专业变化
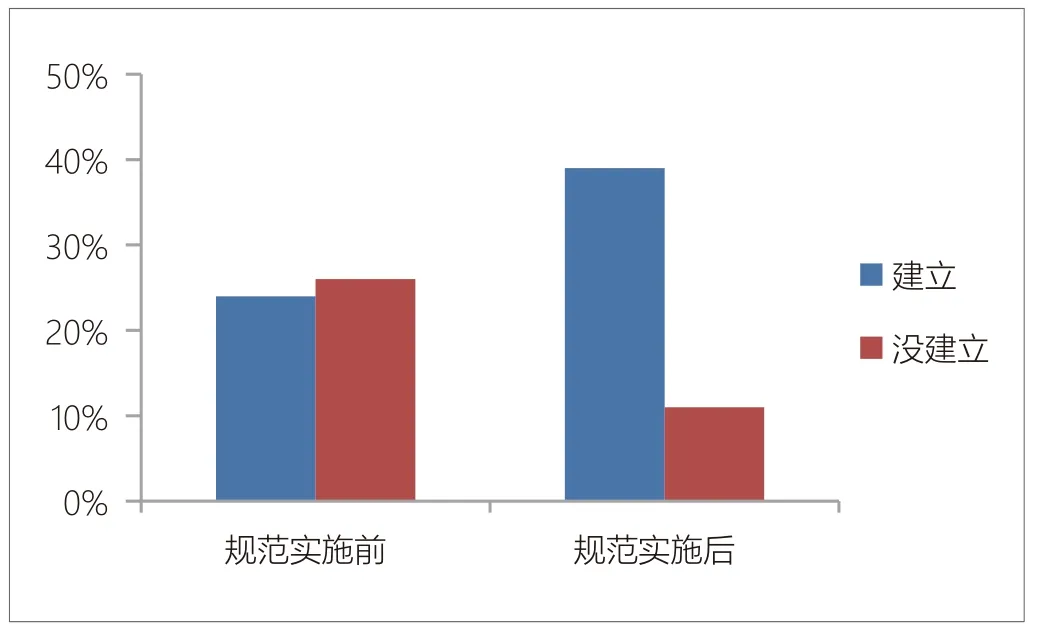
图2. 规范实施前后医疗器械生产企业建立医疗器械临床试验标准操作规程的变化
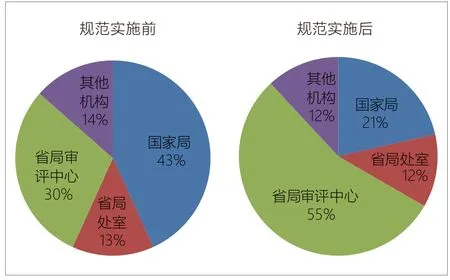
图3. 医疗器械生产企业参加临床试验相关培训的分布
新规范实施后,申办方参加医疗器械临床试验相关法律法规培训的数量由规范实施前的44%增长到60%。主要的培训来源于国家局相关单位和省局,见图3。
2.存在问题
2.1 申办方的医疗器械临床试验团队仍需扩大
虽然临床试验团队相对新规范实施前有所壮大,但申办方的医疗器械临床试验团队仍然相对较小,10人以上的临床试验团队非常少,仅占12%;5~10人的中型团队也仅占14%,而不足5人的小团队占了74%,占绝大多数。从图4可以看出,山东省医疗器械生产企业的临床试验团队规模有待于进一步扩大来满足《医疗器械临床试验质量管理规范》的要求,进一步提升临床试验质量。

图4. 医疗器械生产企业临床试验团队人数
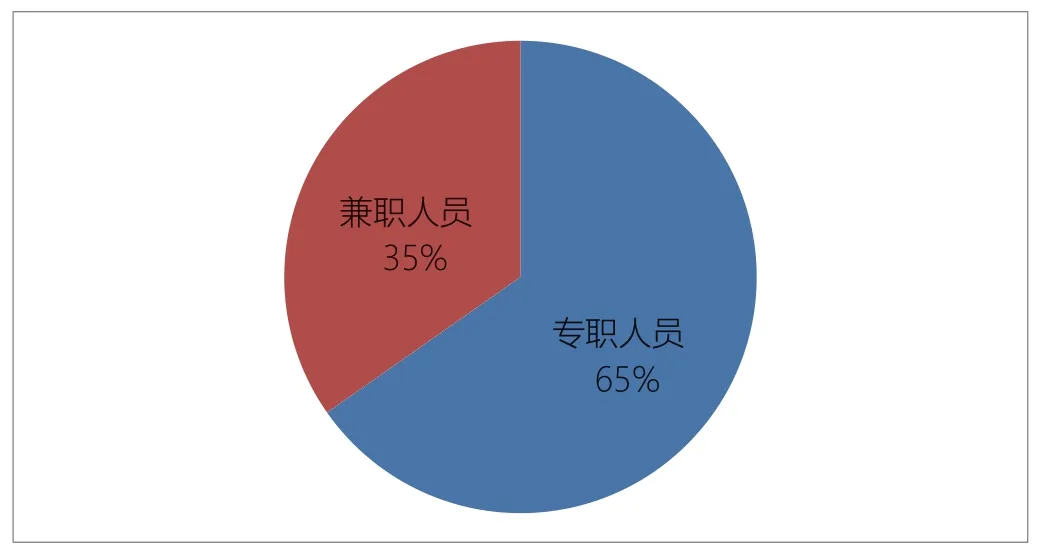
图5. 医疗器械生产企业临床试验专职人员占比
2.2 申办方的临床试验团队专职化程度仍需提高
目前申办方的临床试验团队兼职人员仍占三分之一以上,见图5。兼职人员的其他工作主要有注册、研发、检验、质量管理等。兼职人员无法将全部精力投入到临床试验的实施的各个环节中,专职临床试验团队仍需不断完善。
2.3 目前申办方对医疗器械临床试验的实施需要加强质量控制和专业培训
约69%的申办方选择委托CRO进行临床试验,其中有43%的申办方对于委托的CRO的试验质量基本未进行质控。申办方临床试验团队人员符合法规要求CRA专业的数量仅占约60%。目前仍有40%申办方未按规范要求建立的医疗器械临床试验监查标准操作规程。企业需要加强医疗器械临床试验相关法律法规的学习,目前约有一半的企业未参加过《医疗器械临床试验质量管理规范》的培训。
3.申办方对医疗器械临床试验存在的疑问与困难
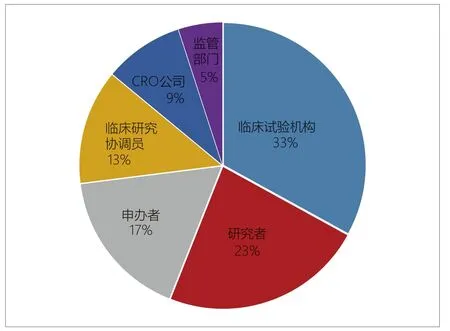
图6. 医疗器械生产企业认为关键参与方的占比
根据与企业的沟通和反馈的调研问卷,显示企业对医疗器械临床试验还存在的疑问与困难如下:①选择临床试验机构困难,导致全国范围选机构,选择周期长。②医院伦理委员会召开会议周期太长,且审核更加严格,准备各中心伦理会以及各临床药理机构合同所需时间延长,导致很多医疗器械临床试验项目伦理批件不能在允许的时间内完成备案,只能做伦理批件延长。③对《医疗器械临床试验质量管理规范》中的某些内容理解不到位,只能咨询相关单位,并参加培训班进行相关规范及法规的学习。④对医疗器械临床试验的实施过程不了解,具体细节做的不到位。⑤对于《医疗器械临床试验现场检查要点》存在某些疑惑,申办方认为目前国家食药总局及省局检查体外诊断试剂临床试验检查要点中对于体外诊断试剂的管理、样本的管理等均有检查要求,但体外诊断试剂临床试验没有明确的临床试验管理规范出台,体外诊断试剂临床试验实施过程中遵循的法规不明确,仅有体外诊断试剂临床试验指导原则,而体外诊断试剂临床试验指导原则并未对外诊断试剂的管理、样本的管理等作详细要求。因此试验过程中对于临床试验用体外诊断试剂的管理、样本的管理、知情同意情况没有统一的标准来遵循。
4.申办方对医疗器械临床试验提出的意见与建议
针对这些疑问与困难及企业临床试验的现状,企业提出如下建议及意见:①申办方认为CRO、临床试验机构和研究者同样也是临床试验的责任主体,应该有更多要求来约束CRO及临床试验机构和研究者,并且能够调动临床试验机构和研究者的积极性。见图6。②有申办方建议无需所有参与的临床试验单位都要经过伦理委员会同意。因为体外诊断试剂产品,本身就是不需要与患者直接接触,不向患者提供检测报告,且检测结果只是用于对比研究,临床试验对受试者几乎没有任何风险。现在要求每一家临床试验单位都要经过伦理委员会同意,在实际操作过程中会增加很大难度和时间。③各机构伦理委员会对体外诊断试剂临床试验是否需要签署知情同意书的意见不同。希望能够统一尺度。④关于严重不良医学事件(SAE)的申报,申办方建议专门开设一个系统或者是一个公共邮箱来申报。因SAE的申报有24h的时间限制,且现在为了证明已经申报SAE,很多药物临床试验机构要求提供SAE申报的传真回复,如遇到传真机故障或无回复功能的传真机,可能会出现无法及时申报SAE的问题。
5.讨论
国内药品临床试验管理有20多年的历史,医疗器械临床试验检查才刚刚开始,相关要求突然拔高而申办方接受的培训又跟不上,同时,器械的临床试验费用比药品临床试验费用低,尤其对于风险大的医疗器械产品医疗机构基本不愿承接,例如脑内植入物等,监管部门检查增多,导致很多医疗机构合作性差,研究者越明白临床试验的风险,越不愿承接临床试验,因此导致了医疗器械临床试验不如药品的临床试验规范。但通过调研发现,通过国家局、省局等监管部门的有效监管,医疗机构、申办方及CRO的共同努力,使得医疗器械临床试验在逐步规范,目前省内医疗器械生产企业基本能够执行《规范》。《规范》实施后更加注重临床试验队伍建设,临床试验团队更加壮大且专业化程度提高,同时,更加重视对医疗器械临床试验相关法律、法规的培训,对临床监查员专业要求更加严格。另一方面,申办方临床试验团队质量有待提升,临床试验主体责任意识有待加强,在临床试验过程中监查工作是保证整个临床试验过程规范数据结论真实可靠的重要手段之一。因此,申办方需要根据临床试验的复杂程度和参与试验的临床试验机构数目决定监查员人数以及监查的次数,选择符合要求的监查员履行监查职责。监查员要严格遵循由申办者制定的试验用医疗器械临床试验监查标准操作规程,督促临床试验按照方案实施。从医疗器械临床试验前、临床试验进行中、临床试验结束后3个阶段全面的对医疗器械临床试验的进行监查工作。另外需要提高检查一致性,对于检查员、研究者和申办方对检查要点的理解需要进一步提升,医疗器械临床试验检查按检查要点进行检查,很多研究者及申办方对于《医疗器械临床试验现场检查要点》中试验器械的管理、样本的管理等理解不到位,但体外诊断试剂临床试验没有明确的临床试验管理规范遵循,仅有体外诊断试剂临床试验指导原则,而体外诊断试剂临床试验指导原则并未对外诊断试剂的管理、样本的管理等作详细要求。因此,下一步希望可以统一临床试验过程中对于临床试验用体外诊断试剂的管理、样本的管理、知情同意的要求等,尤其对于不同样本的管理及不同试验器械的管理可以出台具体相应的指导意见。
猜你喜欢
现代仪器与医疗(2022年1期)2022-04-19
海外星云(2021年9期)2021-10-14
现代仪器与医疗(2021年1期)2021-06-09
生物医学工程学进展(2021年3期)2021-01-20
大众健康(2020年7期)2020-08-25
天津科技(2020年7期)2020-07-31
质量安全与检验检测(2019年3期)2019-07-31
质量安全与检验检测(2018年6期)2018-12-28
商品与质量(2018年43期)2018-12-06
现代企业文化·理论版(2016年23期)2017-04-01
