
纳米明胶硅氧烷的制备及其负载p53基因对肝癌抑制效果
2018-08-24 01:37,,,,,
浙江理工大学学报(自然科学版) 2018年5期
,,, ,,
(浙江理工大学,a.生命科学学院;b.材料与纺织学院,杭州 310018)
0 引 言
原发性肝癌(Hepatocellular carcinoma,HCC)是一种高度恶性肿瘤,是世界上第五大常见癌症,也是世界上癌症死亡率较高的主要疾病之一[1],在发展中国家,HCC发生率超过80%并在男性发病率中居第二位[2]。目前,治疗肝癌有许多方法,包括手术切除、化疗、局部消融和肝移植,但大多患者发现时已发展成为晚期不适合手术,且这些方法也存在显著的毒性和高复发风险[3-5]。目前在大多数国家中仍缺乏有效的治疗方法,HCC的死亡率几乎等于发病率[6]。因此,开发一种在提高肝癌患者预后能力和尽量减少与治疗相关的毒性的治疗方法是非常迫切的。
基因治疗作为一种新兴的治疗方法,是依赖某种载体递送核酸如质粒DNA、siRNA和miRNA等进入患者细胞从而达到治疗目的[7],现已被广泛认为是治疗许多疾病的有效方法,如心血管[8]、遗传性疾病[9]、神经性疾病[10]、恶性肿瘤[11]以及多基因诱导疾病如血友病[12]和肌肉萎缩症[13]等。迄今为止,近2600个基因治疗临床试验已经完成并在全球获得批准[14]。近年来,随着分子生物学的快速发展以及人类基因组工程的完成,人类疾病DNA基因组学的深入研究,通过基因治疗手段来治愈疾病的新模式策略也越来越为人所接受,但选择一种高效、安全的基因载体和治疗基因仍然是成功基因治疗的巨大挑战。
p53蛋白作为肿瘤抑制因子可参与多种细胞反应,在调节细胞周期阻滞、细胞凋亡、DNA修复、自噬、代谢、mRNA翻译和反馈机制等方面起着重要作用[15]。p53基因的突变在肿瘤的发生发展中起着重要作用,超过50%的癌症(包括HCC)是由p53基因突变引起的[16]。用腺病毒作为基因载体重新引入野生型p53基因已经达到临床水平,其中也包括对HCC的治疗[17-18]。脂质体[15]和阳离子聚合物[19]的非病毒载体也越来越多地被用于p53基因传递治疗HCC,引入野生型p53诱导肿瘤细胞发生凋亡而成为癌症基因治疗的一种新手段。
明胶硅氧烷纳米颗粒(Gelatin siloxane nanoparticles,GS NPs)具有低毒性、可降解性、表面易吸附、易修饰性以及易重复合成等优点,作为生物材料已被广泛用于骨组织工程[20]、脑疾病治疗[21-22]和基因药物载体[23-24]等研究。由于GS NPs表面带有较大量的正电荷,可作为一种潜在的基因载体材料,介导目的基因进入细胞并表达,但对于GS NPs负载治疗基因对癌细胞的效果却鲜有报道。本文采用溶胶-凝胶法探究不同的pH值盐酸溶液对纳米明胶硅氧烷制备的影响及其作为基因载体负载p53基因对肝癌细胞的抑制效果。
1 材料与方法
1.1 材料与仪器
a) 材料:pEGFP-C1-p53(p53)质粒为本实验室所保存,明胶(100 g,美国BBI公司),GPSM(3-缩水甘油醚氧基丙基三甲氧基硅烷,分析纯,美国ACROS ORGANICS公司)、APTMS((3-氨丙基)三甲氧基硅烷,分析纯,美国ACROS ORGANICS公司),FITC(异硫氰酸荧光素,10 mg,美国Sigma公司),CCK-8试剂盒、MTT粉末、PI(碘化丙啶)粉末、RIPA裂解液、SDS-PAGE凝胶配制试剂盒(均购自上海碧云天公司),LysoTracker Blue DND-22、BCA试剂盒(美国Thermo Fisher公司),p53抗体(0.1 mL,美国Novus公司),β-actin抗体(0.1 mL,美国Affinity公司),鼠二抗、ECL试剂盒(杭州联科生物技术股份有限公司)。
b) 仪器:场发射扫描电镜(ZEISS-ULTRA55,日本Hitachi公司),动态光散射仪(LB-550 V,英国Malvern公司),数显控温磁力搅拌器(78HW-3,杭州仪表电机有限公司),凝胶成像仪(GGM/D2,GeneGenius公司),酶标仪(ELx 800,BioTek公司),激光共聚焦显微镜(LSM710 3-channel,德国Zeiss公司)。
1.2 不同pH值条件下纳米明胶硅氧烷的制备及表征
首先分别向20.0 mL的pH值2.0、3.0、4.0和5.0盐酸溶液中加入0.15 g的明胶,40 ℃下加热溶解,配成明胶溶液为0.75%。再向明胶溶液中分别加入0.20 g GPSM(185 μL),于60 ℃,500 r/min的磁力恒温搅拌器上搅拌30 min。然后向反应体系中加入0.08 g APTMS(75 μL),于60 ℃下继续搅拌8~10 h。最后停止搅拌,以14000 r/min,20 ℃离心20 min,获取白色沉淀,超声分散水洗三遍后,以10.0 mL的ddH2O超声波重新悬浮,即得到约4 mg/mL GS NPs悬浮液。
用场发射扫描电镜(FE-SEM)对颗粒形貌进行观察,动态光散射仪(DLS)分析其粒径和表面Zeta电位。
1.3 纳米明胶硅氧烷对p53基因的包封性
选取pH值3.0盐酸溶液为反应基础,新鲜制备好的GS NPs重悬超声分散,浓度为4 mg/mL,分别与pEGFP-C1-p53以10∶1、30∶1、50∶1、100∶1、150∶1和200∶1的质量比均匀混合,用ddH2O补齐体系,涡旋15 s,室温孵育静置60 min,即得到GS-p53纳米复合物。采用琼脂糖凝胶电泳法检测GS-p53复合物中是否有pDNA溢出,评价纳米颗粒对p53的包封效率。
1.4 纳米复合物GS-p53的粒径和表面Zeta电位分析
取新鲜制备的GS-p53纳米复合物悬液,用ddH2O稀释后加入1.2 mL至样品池中,置于动态光散射仪上测定其粒径和Zeta电位。
1.5 对p53基因的释放性
按1.3中方法制备复合物悬液(质量比为200∶1),分成3等份,离心分离(14 000 r/min,20 ℃,20 min)后,弃上清液,分别超声重悬于相同体积的50、150 mmol/L和300 mmol/L的NaCl溶液中,并均置于200 r/min,37 ℃的摇床中振荡30、60 min和120 min后,每组取出等体积的复合物悬浮液离心分离,进行凝胶电泳检测上清液中是否有p53溢出。
1.6 纳米明胶硅氧烷的生物相容性
本文采用CCK-8试剂盒检测GS NPs对肝细胞的毒性。纳米颗粒的无菌处理:将新鲜制备的GS NPs,14 000 r/min,20 ℃,离心20 min后弃上清,75%酒精超声重悬至相同浓度,消毒30 min后,离心分离(14 000 r/min,20 ℃,20 min),用无菌水水洗三次,超声分散到无血清培养基中,得到1 mg/mL的使用液,使用时根据实验浓度要求稀释。
取对数生长期的人肝正常细胞L-02以每孔100 μL,细胞密度为1×105个/mL接种于96孔板中,并放置于37 ℃的5% CO2培养箱中培养。待细胞贴壁生长良好后更换为含有不同浓度(0、0.1、0.2、0.4、0.6、0.8、1.0 mg/mL)GS NPs的无血清培养基,其中每个浓度设5个复孔。分别共培养24、48 h和72 h后,吸除旧培养液并用PBS洗两次,加入100 μL新鲜培养基。避光加入10 μL CCK-8溶液,继续孵育1~2 h后,酶标仪测定波长在450 nm处的各孔吸光值(OD值),并根据式(1)计算细胞存活率:
(1)
其中:X为细胞存活率;OD2为实验组的OD值;OD1为对照组的OD值;OD0为Blank组的OD值。
1.7 GS-p53细胞内摄取情况
荧光标记的纳米颗粒(FITC-GS NPs)的制备:取新鲜制备的GS NPs重悬于pH 值8.0的磷酸缓冲液中配成2~5 mg/mL的浓度。以GS NPs/FITC的比例为300∶1加入FITC溶液,避光室温振荡反应1~2 h。14,000 r/min,20 ℃离心20 min后,弃上清,ddH2O超声洗涤三次,得到FITC-GS NPs。荧光标记的FITC-GS/p53-PI纳米复合物的制备:先将p53用PI进行标记,将质粒p53与PI以1∶1比例混匀,室温避光孵育20 min后,即得到PI-p53质粒。然后将FITC-GS NPs与PI-p53质粒按1.3中的方法混合后离心弃上清(质量比为200∶1),即得到FITC-GS/p53-PI纳米复合物。激光共聚焦样品制备:取1.0 mL对数生长期、5×104个/mL的Hep-3b细胞接种于35 mm玻底培养皿(共聚焦专用培养皿)中,待细胞贴壁生长良好后,PBS洗涤两次,更换为900 μL无血清DMEM培养基,避光加入100 μL新鲜制备、2 mg/mL的FITC-GS/p53-PI纳米复合物悬液,分别共培养6 h和24 h。吸去旧培养液,PBS清洗两次。更换为1.0 mL新鲜DMEM培养基,加入50 μL 1 μmol/L LysoTracker Blue(标记溶酶体)染色液继续孵育2 h。去除旧培养基,PBS洗涤三次,每次清洗3~5 min。加入1.0 mL 4%多聚甲醛室温固定15 min。去除固定液,PBS洗涤三次。于玻底滴加一滴抗荧光淬灭封片剂,4 ℃黑暗环境下短时间保存。所有操作均需避光。激光共聚焦显微镜下观察:绿色代表GS NPs,红色代表p53质粒,蓝色代表溶酶体细胞器。
1.8 GS-p53对肝癌细胞的抑制效果
本文采用MTT法检测GS-p53处理后,肝癌细胞的存活率情况。取生长状态良好的肝癌细胞Hep-3b消化计数后调整细胞密度为1×105个/mL,以每孔100 μL接种于96孔板,培养24 h后,弃掉旧培液,加入不同处理组的无血清培养液:裸p53、GS NPs、GS-p53(每孔纳米颗粒的终浓度为600 μg/mL,p53浓度3 μg/mL),继续共培养24、48 h后,PBS清洗两次,更换新鲜培养液,每孔加入20 μL浓度为5 mg/mL MTT溶液,培养箱内继续孵育4 h后,小心吸除每孔内培养基,加入150 μL DMSO,轻微振荡10 min,酶标仪测定490 nm处的吸光值。计算细胞存活率。
1.9 Western blot检测p53蛋白表达水平
GFP-p53融合蛋白的收集:取对数生长期Hep-3b细胞以每孔2.0 mL培养基和2×105个细胞接种于六孔培养板,待细胞贴壁良好后,更换为无血清DMEM培养基,分别加入等量的p53和GS-p53(纳米颗粒终浓度为1 mg/mL),于37 ℃,5% CO2培养箱中共培养48 h后,弃尽旧培养液并用PBS清洗两次,每孔加入100 μL RIPA裂解液,冰上充分裂解30 min后收集并加入6×loading buffer于100 ℃金属浴10 min,用于SDS-PAGE电泳或-20 ℃短期保存。
SDS-PAGE电泳:本文采用12%的分离胶和5%的浓缩胶,按试剂盒操作说明配制SDS-PAGE。每孔加入50 μg蛋白样品,Maker 5 μL,于80 V电压下电泳至浓缩胶分离胶分界处时,加大电压至120 V,电泳约1.5 h。转膜:电泳结束后以负胶正膜(负极-海绵-滤纸-凝胶-PVDF膜(甲醇活化20 s,正面朝下)-滤纸-海绵-正极)的顺序组装好并置入电转槽中,加入4 ℃预冷的转膜缓冲液,于冰上转膜,电压100 V,时间90 min。封闭:转膜结束后,TBST清洗三次,每次5 min,置于5%脱脂奶粉封闭液中,119 r/min室温封闭2 h。抗体孵育并显影:弃掉封闭液,加入10.0 mL一抗溶液(p53抗体以1∶1000比例稀释,内参β-actin以1∶3000比例稀释),119 r/min 4 ℃孵育过夜。TBST清洗四次,每次15 min,加入鼠二抗溶液(以1∶5000比例稀释),119 r/min室温孵育2 h。TBST清洗四次后置于超灵敏化学发光成像仪中曝光检测。
2 实验结果与分析
2.1 不同pH值条件下的GS NPs制备
为了探究溶胶-凝胶法制备GS NPs过程中盐酸溶液pH值的影响,本实验以不同pH值盐酸溶液为反应基础,分别研究pH值2.0、3.0、4.0和5.0盐酸溶液条件对GS NPs粒径、表面Zeta电位和形貌的影响,结果如表1和图1所示。表1结果显示:pH值2.0盐酸溶液中,反应难以形成乳白色悬浊液即GS NPs;当在pH值为3.0、4.0和5.0盐酸溶液中,反应8~10 h均可得到乳白色悬浊液,因此采用溶胶-凝胶法合成GS NPs过程中,必须在pH值大于2.0的弱酸环境中进行,GPSM中环氧基团才能和明胶的氨基反应以形成明胶-GPSM化合物。随着pH值的增大,纳米颗粒的平均粒径由255 nm增大到316 nm,分散性系数由0.21增大到0.31,粒径分布如图1所示:pH值越小,粒径分布越均匀,pH值3.0和4.0盐酸条件下主要分布在200~400 nm,pH值5.0时分布较宽;Zeta电位并未因粒径分布发生较大变化,稳定在37~40 mV(表1)。以上结果表明,当其他反应条件不变时,盐酸溶液的酸性越弱,制备的GS NPs平均粒径越大,分布越宽,而对表面组成无较大影响,仍带正电荷。
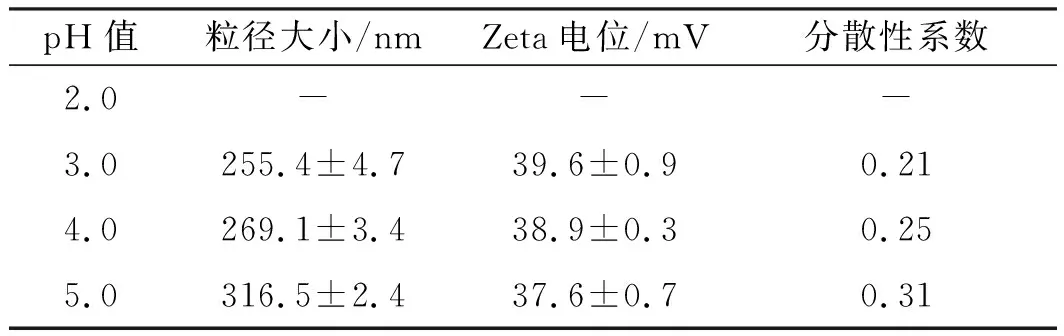
表1 不同pH值体系下GS NPs的粒径、Zeta电位和分散性系数
注:pH值2.0盐酸溶液下,未形成乳白色悬浊液即GS NPs。

图1 不同pH值盐酸溶液下GS NPs的粒径分布
图2为不同pH值盐酸溶液下GS NPs的场发射扫描电镜图。图2(a)-(c)显示,pH值大于2.0盐酸环境下制备的GS NPs呈不规则的球形形貌,分散性较好,存在一种粒径均一约50~100 nm的小颗粒。pH值越大,纳米颗粒粒径越大,盐酸溶液pH值5.0时形成的粒径约300 nm以上明显比pH值3.0时大,pH值3.0和pH值4.0时形成的GS NPs粒径较均一,约250~300 nm间,且pH值3.0下形成的较小颗粒相对于pH值4.0较多,可能导致pH值3.0平均粒径相对较小。与粒径DLS结果综合分析,pH值3.0盐酸溶液下制备的GS NPs分散性较好,平均粒径较小,粒径分布集中,表面带有一定的正电位。

图2 不同pH值盐酸溶液下GS NPs的FE-SEM图
2.2 纳米明胶硅氧烷对p53基因的包封性
为了定性分析GS NPs对pEGFP-C1-p53的包裹能力,对纳米复合物进行凝胶电泳检测,结果如图3所示,随着质量比的逐渐增大,条带亮度逐渐减弱,点样孔的亮度逐渐增强,表明游离的p53逐渐减少,而在点样孔中滞留的p53逐渐增多,说明纳米颗粒的包裹能力随着其含量的增加而增强。其中GS NPs在质量比为30∶1时,未出现明显条带,表明GS NPs以30∶1的质量比能很好地包裹住p53。

图3 GS NPs对pEGFP-C1-p53的包封性检测
注:泳道ND为裸p53;泳道1~6分别为GS NPs/p53质量比为10∶1、30∶1、50∶1、100∶1、150∶1、200∶1。
2.3 GS-p53的粒径和表面Zeta电位
对于非病毒基因载体来说,其粒径和表面电位是影响细胞内化效率和进入细胞方式的重要影响因素[25]。通过对GS-p53纳米复合物的粒径和表面电位分析,分析其最适进入细胞的质量比复合,结果如图4所示。图4表明:GS NPs与p53复合后,表面电位呈现明显的下降,其原因是纳米颗粒负载p53质粒是通过正负电荷静电吸附作用使pDNA结合到纳米颗粒表面,屏蔽纳米颗粒表面的正电荷,导致了其表面电位的下降。GS NPs粒径明显呈现先增后降的趋势,当复合物表面电位为0左右,粒径达到微米以上,说明复合物呈现明显的团聚现象,此时质量比30∶1,结合图3中的GS NPs的包封性,推测其原因可能是当质量比为30∶1时,GS NPs刚好能最大比包裹住质粒p53,从而导致复合物呈电中性,复合物间的同种电荷排斥力减弱,而呈现明显的团聚。由图4还可知,随着纳米颗粒质量比的增加,复合物表面电位大体呈现逐渐增大趋势,最后趋于稳定,这表明随着纳米粒子的增多,其吸附能力越强,当达到一定包裹比例后,pDNA含量不会显著影响其表面电位。而当复合物表面电位稳定于30 mV左右时,其粒径仍有一定程度的增大,但总体小于400 nm,表明粒径的增大主要是由于pDNA结合到纳米颗粒表面引起的。纳米颗粒进入细胞主要是由于表面正电荷与细胞膜表面带负电荷分子的静电相互作用而使其与膜结合,因此对于纳米颗粒复合物来说,维持一定的正电荷是很有必要的,且在纳米颗粒与p53质量比为200∶1的条件下GS-p53具有较小的粒径和一定的正电位,因此在后续研究中,GS NPs按此质量比条件包裹p53质粒,作为纳米复合物。

图4 以不同质量比复合的GS-p53纳米复合物粒径和表面电位分析
2.4 GS-p53体外对p53基因的释放性
图5为GS-p53纳米复合物在50、150 mmol/L和300 mmol/L NaCl溶液中体外对p53基因的释放性。图5显示:120 min时,随着NaCl浓度的增加,p53电泳带的亮度逐渐增加,表明p53释放量的增加。纳米复合物在300 mmol/L的NaCl溶液中有明显的p53电泳带,且随着振荡时间的延长,条带亮度增加,表明对p53的释放作用增强。其原因可能是盐溶液环境中,纳米颗粒对p53的静电作用被盐离子竞争性破坏,因此随着盐浓度的增加和时间的延长,离子竞争效应增强,从而导致p53释放增大。

图5 GS-p53对p53的释放性
注:泳道ND为裸p53;泳道1—9分别代表GS-p53纳米复合物在50 mmol/L NaCl溶液中37 ℃恒温振荡30、60 min和120 min(泳道1、2、3),150 mmol/L NaCl溶液中37 ℃振荡30、60 min和120 min(泳道4、5、6),300 mmol/L NaCl溶液中37 ℃振荡30、60 min和120 min(泳道7、8、9)。
2.5 纳米明胶硅氧烷的生物相容性
为了进一步研究纳米明胶硅氧烷作为基因载体的可行性,对其生物相容性进行检测,图6为GS NPs在0.1~1.0 mg/mL范围内,不同处理时间对肝正常细胞L-02的细胞毒性结果。图6显示:GS NPs对肝细胞的毒性随浓度的增加和处理时间的延长,细胞生长受抑制比较明显,表现出轻微的细胞毒性,当浓度小于0.6 mg/mL时,其细胞存活率仍达到80%以上,总体而言,所制备的GS NPs具有良好的生物相容性,具备一个基因载体所必有的生物安全性,为其后续抑癌效果研究奠定基础。

图6 GS NPs的生物相容性
2.6 GS-p53细胞内摄取研究
为探究纳米明胶硅氧烷作为基因载体携载pDNA进入细胞后在胞内的摄取情况,本文通过FITC标记GS NPs,PI标记p53,共培养6 h和24 h后,对溶酶体进行染色,激光共聚焦观察,结果如图7所示。由图7可知,当共培养6 h时,GS-p53荧光点基本与溶酶体荧光点重叠,表明GS NPs携载p53进入细胞后首先被溶酶体所吞噬。24 h后,有少量荧光点(见图7中的Merge图)单独存在,表明随着共培养时间的延长,少部分GS NPs携载p53开始逃逸出溶酶体的吞噬进入细胞质及细胞核中。以上结果表明GS NPs作为基因载体进入细胞后对溶酶体的逃逸情况存在一定的时间依懒性。
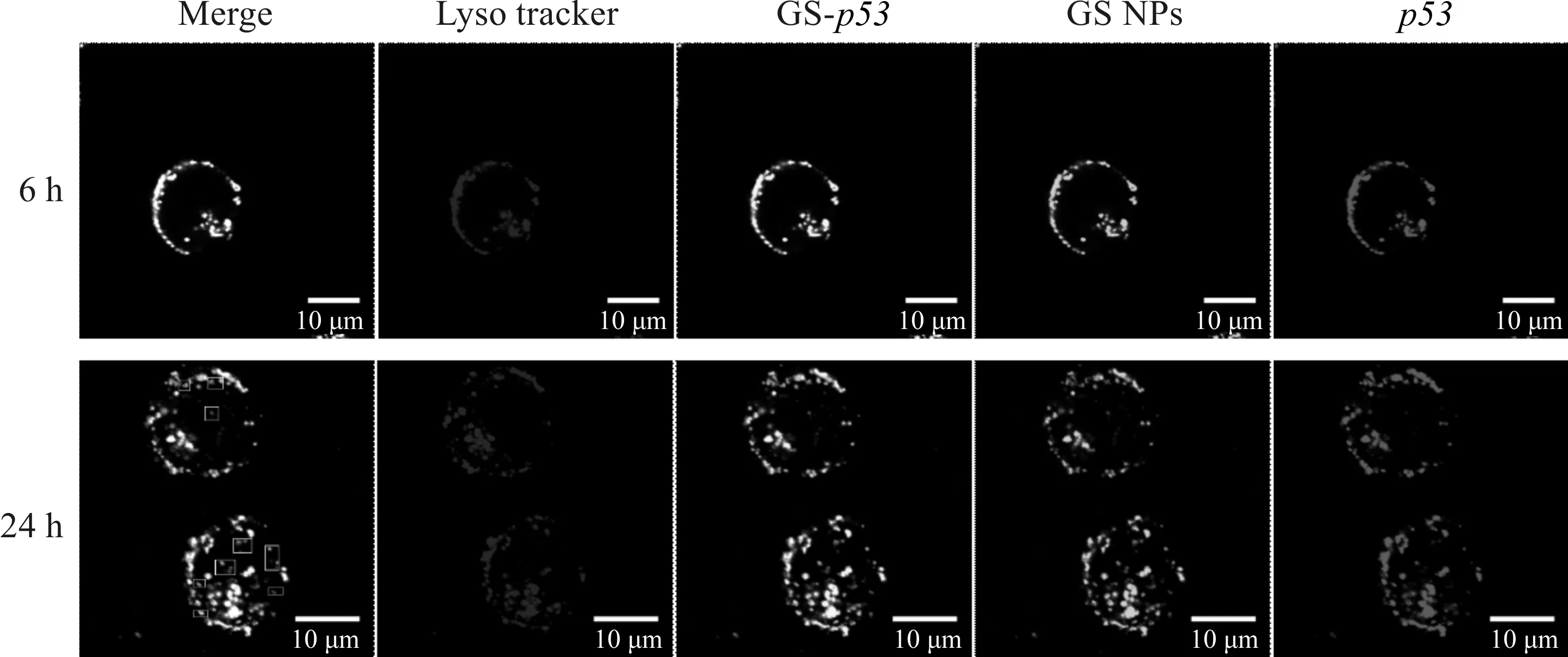
图7 激光共聚焦显微镜观察GS-p53进入Hep-3b细胞后细胞内摄取情况
2.7 GS-p53对肝癌细胞的抑制效果
图8为纳米颗粒终浓度为600 μg/mL(p53终浓度为3 μg/mL)的不同处理组对肝癌细胞Hep-3b分别处理24 h和48 h的抑制效果。从图8中可以看出GS NPs对肝癌细胞具有较好的生物相容性,其细胞存活率达到80%以上。GS NPs携载p53后对肝癌细胞Hep-3b具有较显著的抑制效果;相对于裸p53质粒,GS-p53表现出极显著的抑制作用,具有统计学意义。在培养24 h和48 h后,加入GS-p53纳米复合物组细胞存活率分别为69.7%和63.3%,说明随着处理时间的延长,肝癌细胞的存活率有所降低,在48 h时表现出更强的抑制作用。推测可能是随着时间的延长,更多的GS NPs携载p53逃逸出溶酶体的吞噬,从而进行下一步转染的原因。

图8 GS-p53对肝癌细胞Hep-3b的抑制效果
注:**表示p<0.01,对GS组和GS-p53组进行统计学分析;***表示p<0.001,对pEGFP-C1-p53组和GS-p53组进行统计学分析。
2.8 Western blot检测p53蛋白的表达水平
本文以β-actin蛋白为内参蛋白,用Western blot检测GFP-p53融合蛋白的表达情况,结果如图9所示,由图9可知,内参β-actin蛋白表达水平基本一致,而GFP-p53呈现不同的表达,说明在上样量相同的情况下,对于肝癌细胞Hep-3b,裸p53没有融合蛋白的表达,而在有载体GS NPs的介导下,Hep-3b细胞内能成功表达融合蛋白。因此,GS NPs作为一种基因载体可成功携载p53进入肝癌细胞Hep-3b转染并使GFP-p53融合蛋白表达,从而促进肝癌细胞凋亡,达到一定的抑制效果。

图9 Hep-3b细胞p53蛋白表达检测
3 结 论
本文采用溶胶-凝胶法制备纳米明胶硅氧烷,探讨反应中不同pH值盐酸溶液的影响,并对其进行表征分析;通过凝胶电泳实验检测其对pDNA的负载率及释放性;CCK-8法检测其生物相容性;激光共聚焦观察纳米复合物进入细胞后在胞内的摄取情况;MTT法和Western blot实验综合评估其负载p53基因对肝癌细胞的抑制效果。主要结论如下:
a) 采用溶胶-凝胶法制备GS NPs过程中,必须是在pH值大于2.0的弱酸环境下,反应才能进行从而合成GS NPs;随着pH值的增大,纳米颗粒的平均粒径由255 nm增大到316 nm,粒径分布变宽,但其形貌不会产生较大影响,仍呈无规则球状颗粒,分散性较好,且表面带有一定的正电荷。GS NPs是以pH值3.0的盐酸溶液为反应基础条件下获得,其粒径约250 nm左右、表面Zeta电位38 mV。
b) GS NPs在30∶1时可有效负载p53,但作为基因载体其在200∶1的质量比时合成的纳米复合物GS-p53粒径较小,表面带有一定的正电位,最适合细胞膜的胞吞作用。此外,GS NPs在盐溶液条件下可实现对p53的释放。
c) GS NPs对人正常肝细胞L-02的毒性较低,具有良好的生物相容性。携载p53进入细胞后少部分纳米复合物可逃逸出溶酶体的吞噬,从而进行转染使GFP-p53融合蛋白在肝癌细胞Hep-3b内成功表达,促进肝癌细胞的凋亡,对肝癌细胞具有显著的抑制效果。
猜你喜欢
化工管理(2022年13期)2022-12-02
九江学院学报(自然科学版)(2022年2期)2022-07-02
中国听力语言康复科学杂志(2021年6期)2021-12-21
食品安全导刊(2021年20期)2021-11-28
南昌大学学报(理科版)(2021年3期)2021-10-13
中成药(2019年12期)2020-01-04
安徽化工(2018年4期)2018-09-03
电子制作(2018年9期)2018-08-04
药学研究(2015年11期)2015-12-19
食品工业科技(2014年13期)2014-03-11
