
柱前衍生-超高效液相色谱-串联质谱测定茶叶中草甘膦、草铵膦及主要代谢物氨甲基膦酸残留
2018-09-05 12:54叶美君陆小磊刘相真张海华杜颖颖潘胜东
色谱 2018年9期
叶美君, 陆小磊, 刘相真, 张海华, 杜颖颖, 潘胜东
(1. 中华全国供销合作总社杭州茶叶研究院, 浙江 杭州 310016; 2. 宁波市疾病预防控制中心, 浙江省微量有毒化学物健康风险评估技术研究重点实验室, 浙江 宁波 315010)
由中国农药信息网(http://www.Chinapesticide.gov.cn)和文献[1]可知,草甘膦(glyphosate, GLY)和草铵膦(glufosinate, GLUF)是我国登记的允许茶树使用的除草剂,茶叶中草甘膦和草铵膦的最高残留限量(RML)较低,与日本和欧盟等国家和地区的RML基本相近(中国为1.0 mg/kg和0.5 mg/kg,日本为1.0 mg/kg和0.3 mg/kg,欧盟为2.0 mg/kg和0.1 mg/kg)。茶多酚、氨基酸、咖啡碱、茶多糖等700余种内源性成分赋予茶叶独特的健康保健功能的同时[2],极大地增加了茶叶中草甘膦和草铵膦检测前处理和仪器分析的难度,进而影响样品检测的灵敏度(检出限和定量限)、精密度(重复性)和准确度(回收率)。
草甘膦和草铵膦的检测方法主要包括离子色谱法(IC)、高效液相色谱-荧光法(HPLC-FD)、气相色谱-质谱法(GC-MS)和液相色谱-质谱法(LC-MS)[3-5]。草甘膦和草铵膦及其主要代谢物氨甲基磷酸(minomethylphosphonic acid, AMPA)的分子质量小、极性大、不溶于有机溶剂、水溶性强,与有机物有很强的结合能力,直接分析难度较大[6]。黄嘉乐等[7]采用离子色谱法直接检测茶叶中草甘膦和草铵膦,由于其定量限较高,依据GB 2763-2016《食品安全国家标准 食品中农药最大残留限量》中该除草剂的RML,该检测方法无法满足日常检验要求,因此植物源样品主要采用衍生法间接检测。茶叶中氨基酸种类繁多且含量高,草甘膦和草铵膦属氨基酸类除草剂,该衍生产物与氨基酸衍生产物结构相似,荧光检测器分辨能力低,假阳性高,不利于茶叶样品的准确分析。气相色谱-质谱法(GB/T 23750-2009)衍生试剂七氟丁醇(HFB)价格昂贵,衍生剂三氟乙酸酐(TFAA)易挥发,衍生条件苛刻,日常检验应用较少。9-芴甲基氯甲酸酯(FMOC-Cl)作为氨基酸的保护基团,用于固相多肽合成和有机物合成,也作为衍生试剂常用于氨基酸的定性定量分析[8-11]。Ibáez等[12]、曹赵云等[13]和吴晓刚等[14]将该衍生剂引入氨基酸类除草剂的检测,Ibáez等报道了草甘膦、草铵膦和氨甲基磷酸衍生产物(FMOC-GLY、FMOC-GLUF和FMOC-AMPA)的质谱图,曹赵云等和吴晓刚等阐明该衍生产物在质谱中的裂解途径。由于FMOC-Cl易溶于有机溶剂,不溶于水,而形成的衍生产物FMOC-GLY、FMOC-AMPA和FMOC-GLUF溶于水,基于这一特殊的理化特性,以FMOC-Cl为衍生试剂的柱前衍生-高效液相色谱-质谱法广泛应用于茶叶中草甘膦、草铵膦和氨甲基膦酸的检测[14-21]。
基于FMOC-GLY、FMOC-GLUF和FMOC-AMPA的超高效液相色谱-串联质谱(UPLC-MS/MS)检测技术成熟,实验条件简单,茶叶样品的检测难点主要集中在提取和净化等前处理过程,该过程直接决定茶叶样品中内源性物质的组成,并影响草甘膦、草铵膦和氨甲基膦酸的衍生反应转化率,进而影响样品检测的灵敏度、精密度和准确度。本实验选取水和0.2%(v/v)甲酸水溶液作为提取剂,超声、振荡和旋涡作为提取方式,草甘膦专用柱(Special SPE)、碳十八柱(C18柱)和阳离子交换柱(PCX)作为固相萃取(SPE)的净化小柱,利用正交试验设计方法,优化提取剂、提取方式和固相萃取净化小柱,系统研究提取和净化等前处理过程对茶叶中草甘膦、草铵膦和氨甲基膦酸检测的影响。本文采用UPLC-MS/MS内标法对茶叶中草甘膦、草铵膦和氨甲基膦酸定性定量分析,以灵敏度、精密度和准确度评价方法的适用性。
1 实验部分
1.1 仪器与设备
UPLC/TSQ Quantum Access MAX超高效液相色谱-三重四极杆质谱联用仪(美国Thermo Fisher Scientific公司);分析天平(感量0.000 1 g,瑞士Mettler Toledo公司); SC-3610低速离心机(安徽中科中佳科学仪器有限公司); FJ-12A固相萃取装置(上海京孚仪器有限公司);超纯水系统(成都康宁实验专用纯水设备厂);漩涡混合器(海门其林贝尔仪器制造有限公司)。
1.2 试剂与材料
甲醇(色谱纯,美国Tedia公司);乙酸铵(色谱纯,美国Fluka公司);甲酸、37%盐酸(色谱纯,德国Merck公司);十水四硼酸钠(分析纯,天津市风船化学试剂科技有限公司);磷酸二氢钾(分析纯,华东医药股份有限公司); FMOC-Cl(衍生级,纯度≥ 99.9%,上海安谱科学仪器有限公司);草甘膦水溶液(100 μg/mL,AccuStandard, Inc);草铵膦(纯度99.0%, AccuStandard, Inc);氨甲基膦酸(纯度99.0%, Chem Service); 1,2-14C15N草甘膦(GLY-15N,纯度96%, TRC); FMOC-GLY和FMOC-AMPA水溶液(10 μg/mL,Dr. Ehrenstorfer); FMOC-GLUF(纯度96.2%, Dr. Ehrenstorfer); 100 mg/3 mL草甘膦专用柱(Special SPE,福建昊阳生物科技有限公司); 200 mg/3 mL C18小柱(C18,岛津技迩商贸有限公司); 60 mg/3 mL阳离子交换柱(PCX,博纳艾杰尔科技公司);茶粉(茶叶磨碎样品,按照GB/T 8303-2013制备)。
1.3 标准溶液、硼酸钠溶液和衍生液的配制
标准溶液配制:准确称取10.0 mg(精确至0.1 mg)固体标准物质,分别用超纯水溶解并配制成质量浓度为100 mg/L的标准储备液,于4 ℃冰箱中避光保存。将上述标准储备液和液体标准品逐级稀释,配成单标准溶液或混合标准溶液。
50.0 g/L硼酸钠溶液:称取9.46 g Na2B4O7510H2O,采用超纯水溶解并定容至100 mL。
250 g/L氢氧化钠溶液:称取25.00 g NaOH,采用超纯水溶解并定容至100 mL。
1.00 g/L FMOC-Cl丙酮溶液:称取0.10 g FMOC-Cl,采用丙酮溶解并定容至100 mL。
酸度调节剂:称取16.00 g磷酸二氢钾溶于160 mL水,加入13.4 mL盐酸和40 mL甲醇,超声混合均匀。
1.4 液相色谱-串联质谱条件
色谱柱:Thermo Hypersil GOLD C18(150 mm×2.1 mm, 1.9 μm);流动相A为5 mmol/L乙酸铵水溶液(含0.1%(v/v)甲酸),流动相B为甲醇,梯度洗脱条件见表1;柱温:40 ℃;进样量:2.0 μL。
质谱电离模式:电喷雾离子化(ESI);电离源极性:正离子模式;雾化气:氮气;离子喷雾电压:3 500 V;雾化室温度:120 ℃;离子传输管温度:350 ℃;碰撞气:氩气,0.2 Pa;扫描模式:选择反应监测扫描(SRM)。
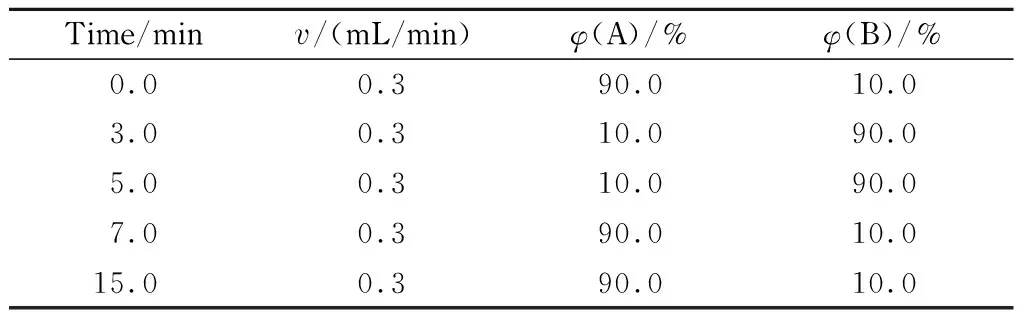
表 1 梯度洗脱程序
A: 5 mmol/L ammonium acetate-0.1% (v/v) formic acid aqueous solution; B: ethanol.
1.5 样品前处理
提取:称取茶粉2.5 g(精确至0.001 g)置于50 mL具塞聚乙烯离心管中,加入10 μg/mL草甘膦同位素内标100 μL,再加入20 mL超纯水,旋涡提取30 min,以2 500 r/min转速离心5 min,取上清液待净化。
净化:分别移取2.0 mL甲醇和2.0 mL 0.5%(v/v)甲酸水溶液依次活化小柱,准确移取1.0 mL提取上清液和100 μL酸度调节剂于10 mL离心管中混合后移取至小柱,再加入1.0 mL 0.5%(v/v)甲酸水溶液洗脱,采用250 g/L NaOH调节流出液至中性并加水定容至3 mL,待衍生。
衍生:取0.6 mL净化液,分别依次加入0.2 mL 50.0 g/L硼酸钠溶液,0.2 mL 1.0 g/L FMOC-Cl丙酮溶液,混匀后于25 ℃下衍生2 h, 0.2 μm有机微孔滤膜过滤,UPLC-MS/MS测定。
2 结果与讨论
2.1 质谱条件优化和衍生产物确证
将FMOC-GLY、FMOC-GLUF和FMOC-AMPA的单标准溶液进质谱仪器直接分析,调节碰撞能量,以获得稳定性好、信号强度高的碎片离子,优化得到的参数见表2。
在25 ℃下,GLY、AMPA和GLUF单标准溶液分别与FMOC-Cl溶液反应2 h,将该衍生反应溶液进质谱分析,谱图显示该衍生反应溶液的离子碎片与FMOC-GLY、FMOC-GLUF和FMOC-AMPA的单标准溶液离子碎片一致。设置液相色谱条件和质谱参数,经UPLC-MS/MS分析,FMOC-GLY、FMOC-GLU和FMOC-AMPA单标准溶液与该衍生反应溶液的液相色谱保留时间一致。由此可见,根据1.5节衍生条件和方法,GLY、GLUF和AMPA与FMOC-Cl反应的产物为FMOC-GLY、FMOC-GLUF和FMOC-AMPA。

表 2 FMOC-GLY、FMOC-GLY-15N、FMOC-GLUF和FMOC-AMPA的质谱参数
* Quantitative ion pair.
2.2 前处理条件优化
2.2.1GLY、GLUF和AMPA在不同固相萃取柱中的洗脱液用量
选取Special SPE、C18和PCX作为茶叶中GLY、GLUF和AMPA检测的净化小柱,以此考察不同固相萃取柱的净化效果。为了确保目标物完全洗脱,首先进行流出液实验,优化各种固相萃取柱的洗脱液用量。以2 mL甲醇和2 mL 0.5%(v/v)甲酸水溶液活化小柱,移取1 mL 100 μg/L的混合标准溶液和100 μL酸度调节剂混合后移取至固相萃取柱,采用0.5%(v/v)甲酸水溶液为洗脱液,每次分别加入0.5 mL洗脱液,洗脱液用量见表3。
根据表3数据显示,以0.5%(v/v)甲酸水溶液为洗脱剂,3种固相萃取柱的洗脱液用量相似,均为1 mL,试样流出液中已存在草甘膦和氨甲基膦酸,前段流出液必须收集。

表 3 目标物在不同固相萃取柱中不同洗脱液用量下的洗脱效果
+: the target compounds were detected in eluate; -: none of target compounds were detected in eluate.
2.2.2提取溶剂、提取方式和固相萃取净化柱的选择
以水和0.2%(v/v)甲酸水溶液作为提取剂,超声、振荡和旋涡作为提取方式,Special SPE、C18和PCX为固相萃取净化小柱,采用正交试验设计研究前处理方法对茶叶中草甘膦、草铵膦和氨甲基膦酸检测的影响,样品添加水平为0.400 mg/kg,实验过程如1.5节所示,正交试验设计见表4,实验结果见表5,色谱图见图1。由图1可知,在相同的色谱条件下,FMOC-GLY、FMOC-GLUF和FMOC-AMPA的保留时间分别约为:FMOC-GLY 4.62 min,FMOC-GLUF 4.91 min, FMOC-AMPA 4.79 min,经Special SPE和C18柱净化,FMOC-GLY和FMOC-GLUF与杂质峰尚未完全分离,且杂质峰响应相对FMOC-GLY和FMOC-GLUF响应较大;经PCX柱净化,FMOC-GLY与杂质峰完全分离,FMOC-GLY和FMOC-GLUF响应相对杂质峰响应较大。

表 4 正交试验设计
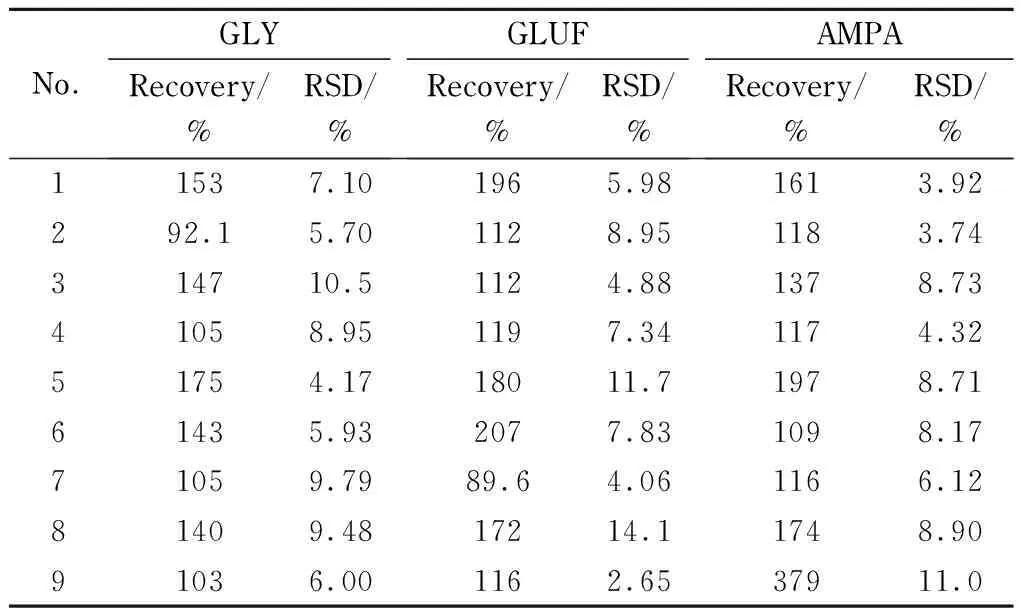
表 5 GLY、GLUF和AMPA在不同前处理方法中的添加回收率和相对标准偏差(n=3)
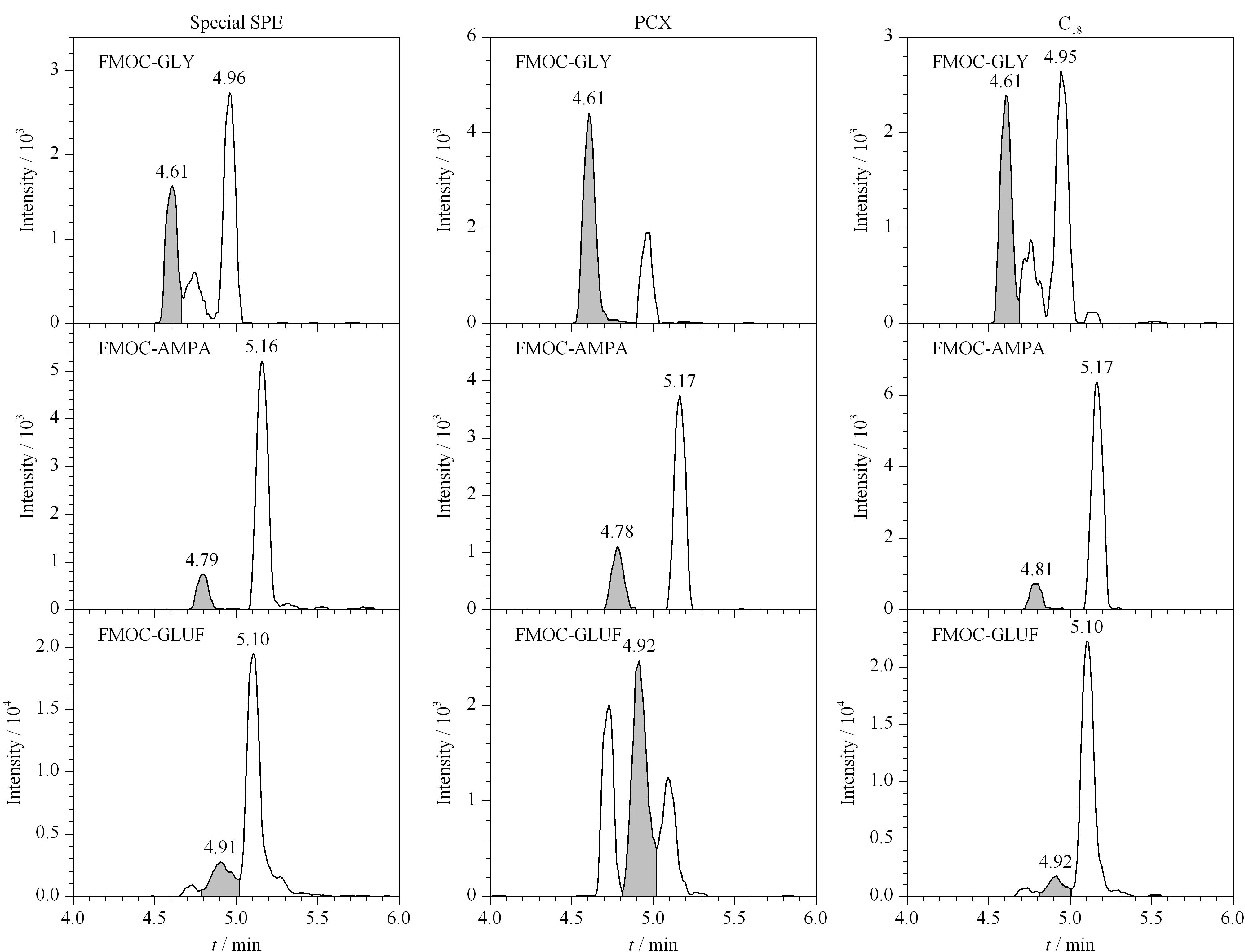
图 1 采用不同固相萃取小柱净化的FMOC-GLY、FMOC-GLUF和FMOC-AMPA色谱图Fig. 1 Chromatograms of FMOC-GLY, FMOC-GLUF, and FMOC-AMPA purified by different SPE columns

图 2 不同实验条件下FMOC-GLY、FMOC-GLUF和 FMOC-AMPA的峰面积Fig. 2 Peak area of FMOC-GLY, FMOC-GLUF, and FMOC- AMPA under different experiment conditions
根据表5可知,No. 2、No. 4和No. 7的回收率为70%~120%,回收率数据尚无显著差异性和规律性,相对标准偏差小于10%,满足检测要求。根据目标物的峰面积(见图2)可知,采用PCX柱的No. 4和Special柱的No. 7, FMOC-GLY和FMOC-GLUF的响应较大,采用C18柱的No. 2, FMOC-AMPA响应较大。
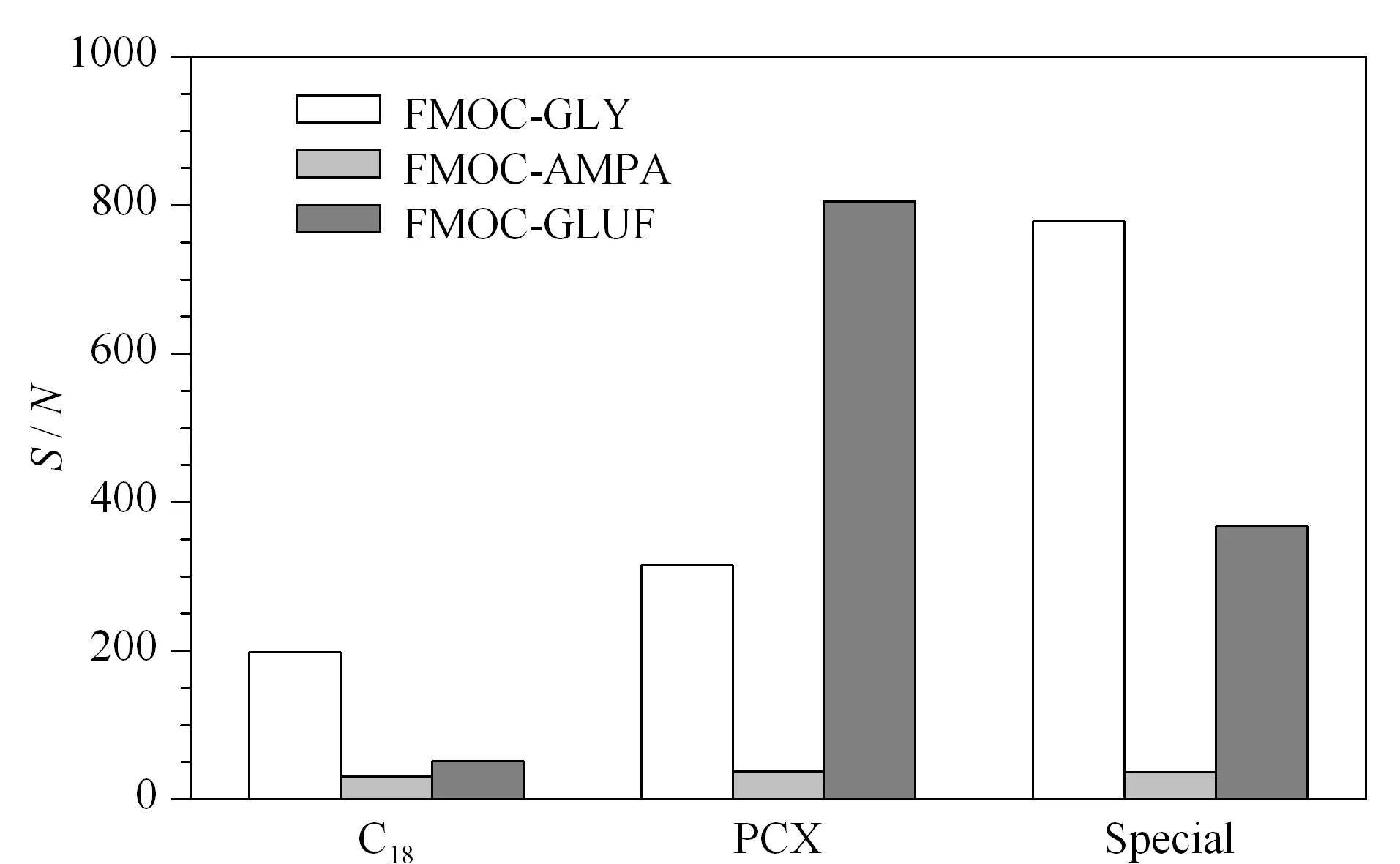
图 3 不同实验条件下FMOC-GLY、FMOC-GLUF和 FMOC-AMPA的信噪比Fig. 3 S/N ratios of FMOC-GLY, FMOC-GLUF, and FMOC- AMPA under different experiment conditions
设计低水平添加实验(在检出限附近),通过信噪比(S/N)进一步确认最优实验方案。将添加水平为0.100 mg/kg的茶叶样品按照No. 2、No. 4和No. 7试验方案和1.5节的前处理过程进行处理,目标物的信噪比见图3。根据图3可知,在0.100 mg/kg添加水平下,采用PCX净化柱的No. 4试验和Special SPE净化的No. 7试验,草甘膦和草铵膦的信噪比较大,氨甲基膦酸在3支净化柱上的信噪比相似。
综合回收率、色谱分离度、目标物响应、杂质干扰和低浓度溶液的信噪比等因素,本实验的最优前处理方法即采用水为提取剂,旋涡为提取方式,PCX柱为净化小柱。
2.3 方法评价
2.3.1标准曲线、基质效应、方法检出限和定量限
分别以PCX柱净化的茶提取液和纯水配制基质标准溶液和溶剂标准溶液,以FMOC-Cl溶液进行衍生,以目标物与草甘膦同位素内标物的峰面积比值作为纵坐标,目标物浓度与草甘膦同位素内标物浓度的比值为横坐标,绘制基质匹配标准曲线和溶剂标准曲线。基质效应以η表示:η=(基质匹配标准曲线的斜率-溶剂标准曲线的斜率)/溶剂标准曲线的斜率[22]。结果表明,在1~100 μg/L范围内线性关系良好,基质匹配标准曲线、相关系数(R2)和基质效应参数见表6。由表6可知,η<10%,说明无明显基质效应,由此可见,草甘膦同位素内标可以大幅度降低基质效应。在空白茶叶样品中添加草甘膦、草铵膦和氨甲基磷酸,并经1.5节样品前处理方法处理,根据3倍信噪比(S/N=3)确定方法的检出限(LOD),根据0.050 0 mg/kg添加水平的回收率和RSD数据确认方法的定量限(LOQ)(见表6)。
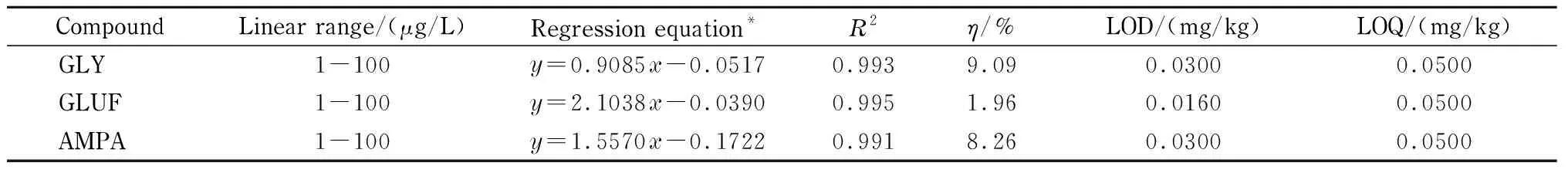
表 6 线性范围、基质匹配线性方程、相关系数、基质效应、检出限和定量限
y: ratio of peak areas of target and internal standard;x: ratio of mass concentrations of target and internal standard.
2.3.2方法准确度和精密度
分别称取2.50 g茶叶空白样品,然后加入适量混合标准溶液,配制成低、中、高3个添加水平(0.050 0、0.400和1.20 mg/kg),按照1.5节样品前处理方法进行提取、净化和衍生,然后采用UPLC-MS/MS检测,每一个加标水平测定6次,结果见表7。

表 7 草甘膦、草铵膦和氨甲基磷酸在茶叶中的添加回收率和RSD(n=6)
由表7可知,草甘膦、草铵膦和氨甲基磷酸在低、中、高3个水平均有着较好的准确度与精密度,符合实验室日常检测要求。
2.3.3与其他方法的比较
目前国内现行标准GB/T 23750-2009《植物性产品中草甘膦残留量的测定 气相色谱-质谱法》、SN/T 1923-2007《进出口食品中草甘膦残留量的检测方法 液相色谱-质谱/质谱法》和SN/T 3983-2014《出口食品中氨基酸类有机磷除草剂残留量的测定 液相色谱-质谱/质谱法》等前处理均采用美国Bio-Rad CAX阳离子交换树脂小柱,按照上述标准方法对茶叶中草甘膦和草铵膦进行检测,经过长期检测发现诸多问题:Bio-Rad CAX为湿柱,柱体积大,吸附容量小,必须采用常压过柱方式,洗脱液用量大且水占比高,不易浓缩蒸干,实际工作中需要耗费大量的时间和精力,而且Bio-Rad CAX阳离子交换树脂小柱价格昂贵。本实验采用PCX干柱,柱体积小,可采用减压过柱方式,采用0.5%(v/v)甲酸水溶液作为洗脱液,洗脱液用量小,在UPLC-MS/MS灵敏度范围内,无需浓缩已达到定量限要求,价格低廉,可弥补Bio-Rad CAX柱的缺点。
2.4 实际样品检测
采用上述方法检测837份绿茶、红茶、青茶、黑茶、白茶、黄茶、代用茶和速溶茶等茶叶样品,其中草甘膦阳性样品29份,残留量在0.140~2.36 mg/kg之间,草铵膦阳性样品2份,残留量分别为0.110 mg/kg和0.130 mg/kg,氨甲基磷酸阳性样品37份,残留量在0.120~2.15 mg/kg之间。草甘膦、草铵膦和氨甲基磷酸的检出率分别为3.46%、0.24%和4.42%。按照GB 2763-2016的草甘膦和草铵膦的MRL判定,茶叶样品草甘膦超标2份,超标率为0.24%,茶叶样品草铵膦未超标。目前GB 2763-2016尚未制订氨甲基磷酸的MRL。
3 结论
本文采用正交试验设计筛选出最优前处理方法,以水为提取剂,旋涡提取,PCX强阳离子交换固相萃取干柱净化,建立了柱前衍生-超高效液相色谱-串联质谱测定茶叶中草甘膦、草铵膦及其主要代谢物氨甲基膦酸残留量的实验方法。该分析方法高效、便捷,价格低廉,可操作性强,方法的灵敏度、精密度和准确度满足茶叶的日常检测要求。根据本实验现象可知,茶叶丰富的内源性物质对草甘膦、草铵膦和氨甲基磷酸检测影响较大,茶叶基质的有效去除是排除假阳性样品的关键。基于这一实验结果,进一步深入探讨茶叶内源性成分对草甘膦等除草剂衍生反应的影响,发现其中的内在规律,可为提取溶剂和提取方式的选择、净化材料的开发和衍生试剂的选取等提供理论和现实依据。
猜你喜欢
世界农药(2022年11期)2022-12-08
少年文艺(2022年8期)2022-07-08
农药科学与管理(2019年6期)2019-11-23
世界农药(2019年2期)2019-01-06
现代农药(2017年2期)2017-04-18
中国经济周刊(2017年6期)2017-03-21
今日农药(2016年7期)2016-05-14
环境科技(2015年4期)2015-11-08
中国医疗美容(2015年1期)2015-07-12
营销界(2015年23期)2015-02-28
