
多特征融合的lncRNA识别与其功能预测
2018-11-05 09:12常征孟军施云生莫冯然
智能系统学报 2018年6期
常征,孟军,施云生,莫冯然
(大连理工大学 计算机科学与技术学院,辽宁 大连 116023)
近年来,非编码RNA(non-coding RNA,ncRNA)识别的相关研究已成为人们关注的热点。一直以来,转录本被大家普遍认为只起到翻译蛋白质的作用,但随着人类基因组注释工作的不断推进,研究结果表明只有大约1%~2%的基因参与了编码蛋白的工作[1],而以往被大家忽略的非编码序列也在整个生命活动中扮演着至关重要的角色。这些非编码序列中,有一种长度大于200 nt、无法编码蛋白质的转录本尤其受到关注,被称为长链非编码RNA(long non-coding RNA,lncRNA)[2]。近年来发现lncRNA具有调节生物体生命活动的重要作用[3-4],而各种传统的实验方法,一方面需要花费大量时间和高额费用,另一方面,因为lncRNA的低表达和低保守性等原因,在识别lncRNA方面受到不同程度的影响。研究人员对人和动物进行了大量的实验,并且出现了具有良好鲁棒性的lncRNA识别软件。
RNAseq和全基因组阵列分析显示,植物体内也存在大量的lncRNA,它们在植物的开花、雄性不育、营养代谢、生物和非生物胁迫等生物过程中起着调节因子的作用[5]。与哺乳动物相比,植物ncRNA的研究起步比较晚,且多数集中在短链非编码RNA上,这为植物lncRNA识别与分析带来了困难。研究植物lncRNA将帮助生命学科的工作者进一步揭示植物内部生命活动,因此深入研究植物lncRNA并预测其功能具有非常重要的意义。
目前,在计算预测lncRNA方面,许多研究工作都利用机器学习算法建立预测模型,通过输入各类序列特征、结构特征,构建识别lncRNA的分类器模型。研究表明,对于lncRNA识别,通过提取开放阅读框、密码子频率偏好性、与已知蛋白质相似度等特征作为输入,对线性回归、支持向量机以及其他模型进行训练得到的分类器具有良好的分类效果[6]。近年来衍生出的预测软件多采用以上特征。其中,CPC[7]和 CPAT[8]都是通过序列特征来区分编码和非编码RNA;CNCI[9]能够将训练好的分类器运用到近亲物种的lncRNA识别;PLEK[10]可以从高通量测序的转录本中识别lncRNA。 然而,大多数软件只在动物数据集上得到良好的验证,专门为植物lncRNA识别设计的软件目前还比较稀缺。
随着基因组学研究的不断深入,产生了大量未被标注的基因序列。由于生物实验方法验证基因功能的代价十分昂贵,如何通过计算机方法对基因序列功能进行大规模预测成了近年来生物信息学的研究热点之一[11]。
为了进一步提高植物lncRNA预测的准确性,基于机器学习分类算法,通过对下载的高可信度数据提取开放阅读框、k核苷酸频率以及二级结构特征等多特征融合[12]作为输入特征,训练朴素贝叶斯、支持向量机和梯度提升决策树3种分类模型,并采用加权投票的多分类器集成方法,集成分类结果以得到更好的分类性能。利用标注测序数据测试集对模型进行验证、分析并选择性能最好的将其作为最终分类器。提出的方法通过五折交叉验证,得到了较好的性能。在功能预测方面,根据lncRNA-microRNA、microRNA-mRNA相互作用关系,建立调控网络,利用相关联的RNA预测lncRNA的功能。
1 基于多特征融合的lncRNA预测
1.1 数据集
拟南芥的生物学实验数据和基因注释信息相对比较丰富,常被广泛用于植物胁迫响应的研究中[13]。本文使用的正集数据为PNRD[14](http://structuralbiology.cau.edu.cn/PNRD/) 2 565条具有高可信度的拟南芥lncRNA序列。负集数据是从Ref-Seq数据库下载的48 148条mRNA序列。为了保证正负样本均衡,从负集原始数据中随机采样出2 500条mRNA作为最终训练集,如表1所示。

表 1 数据集信息Table 1 Dataset information
1.2 开放阅读框
在分子遗传学中,开放阅读框(open reading frame,ORF)是阅读框的一部分,具有潜在的翻译能力[15]。研究表明,mRNA的ORF覆盖率明显高于lncRNA,且mRNA具有更多的完整性开放阅读框[16]。首先利用TransDecoder软件包计算得到每个序列的开放阅读框信息,然后对其分别提取完整性、ORF覆盖率以及归一化的ORF值3种特征。将完整性定义为一个布尔变量bool,0代表不存在完整性ORF,1表示存在完整性ORF。覆盖率Coverage等于所有的合法ORF(本文只考虑正链情况下)的长度与RNA序列长度之比,归一化ORF Normalized_ORF是序列中ORF个数n与RNA序列长度L的比值,分别定义为
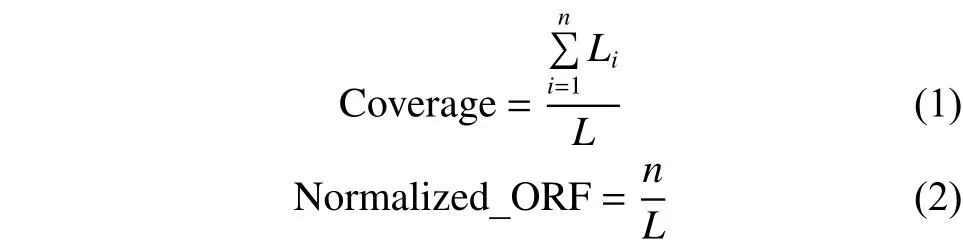
式中Li代表序列中第i个ORF的长度。
整合3种特征得到特征向量:
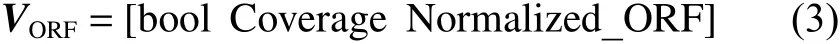
1.3 二级结构
二级结构(secondary structures,SS)是单条序列通过碱基配对自身形成茎区和环球,与RNA的功能息息相关,可以作为识别lncRNA的重要依据。目前,预测二级结构的计算方法分为两类:基于热力学和基于系统发生学。前者认为:生物体在形成高维结构的时候,将使自身达到稳态结构,因此释放的能量应更多。研究表明,二级结构越是稳定(释放的自由能越多)其潜在的编码能力越强。同时,二级结构的稳定性与RNA序列中配对碱基个数以及核苷酸C和G的含量有一定的关系。本文使用ViennaRNA[17]工具包对序列形成二级结构释放的自由能进行计算并得到二级结构的点括号表示形式,然后从中提取出配对碱基的个数以及C和G碱基的含量。归一化最小自由能由如下:

式中:MFE是释放的自由能,L是RNA序列的长度。
整合上述3个特征得到如下特征向量:

式中np为配对碱基的个数。
1.4 k-mers
密码子是遗传物质编码的信息由活细胞转化为蛋白质的一套规则,蛋白质为保证其某些生物功能,在自然选择下会表现出对某些密码子的偏好性。因此mRNA在密码子方面表现出一定的保守性,而不编码蛋白的lncRNA其保守性较差。所以可以使用密码子频率当作识别lncRNA的一个特征。然而,因为无法准确定位mRNA编码区域,且lncRNA有多个编码区域,直接计算密码子频率存在一定困难。为解决以上问题,使用一个近似的解决方法:k-mers特征计算。
一个k-mer具有k个核苷酸,每个核苷酸可以是 A、C、G 或 T,k取值为 1、2和 3,则有 4+16+64=84种模式:4个 1-mer,16个 2-mer,64个3-mer。使用一个长度为k的滑动窗口来匹配上述k-mer。滑动窗口沿RNA序列以步长为1核苷酸进行滑动匹配,使用ci表示匹配到的次数(),并且为每个k-mer分配一个系数wk,从而使得每类频率对预测效果的影响一样,具体如下:

式中:sk是总的匹配次数,L为RNA序列长度,fi为每种k-mer出现的频率,则得到二级结构特征向量:

本文选择融合上述3类特征组成含90维的特征集作为最终的特征向量:

1.5 构建分类模型
朴素贝叶斯方法(Naive Bayes)是基于贝叶斯定理的监督学习算法,即简单地假设每对特征之间相互独立。相比于其他更复杂的方法,朴素贝叶斯学习器和分类器非常快,并且有助于解决高维数据问题。支持向量机是一种基于统计学习的分类方法[18],其模型参数确定会对应到一个凸最优化问题,因此可以保证得到最优解。目前流行的CPC、CNCI等软件都使用SVM作为分类器。梯度提升决策树(gradient boost decision tree,GBDT)是对于任意可微损失函数的提升算法的泛化,它具有强大的预测能力以及在输出空间中对异常点的鲁棒性。
结合训练集高维度、非连续等特征,以及模型自身的鲁棒性,本文选择基于高斯分布的朴素贝叶斯模型、支持向量机以及梯度提升决策树3个模型进行训练。然后使用网格搜索法分别调整3个分类器的超参数。并且采用加权投票分法来融合上述3个分类模型的输出得到最终的预测结果。
1.6 性能评价指标
本文选择使用准确率(Accuracy,ACC)、精确率 (Precison,P)、召回率 (Recall,R)、F1值 (F1_score)来评估训练出的分类模型。定义如下:

式中:TP指将正类预测为正类数,FN指将正类预测为负类数,FP指将负类预测为正类数,TN指将负类预测为负类数。
2 LncRNA功能预测
2.1 数据集
用于构建互作网络的microRNA序列是从miRBase[19](http://www.mirbase.org/index.shtml)下载的427条成熟拟南芥microRNA序列。lncRNA以及mRNA则选用上述下载的具有高可信度的2 565条lncRNA与2 500条mRNA。
2.2 靶向预测提取互作对
研究证明,胁迫作用下,植物的性状将发生改变,而这个过程是由多个基因相互作用形成的,lncRNA也参加其中。作为竞争性内源RNA或者microRNA内源性模拟靶标的lncRNA,可以有效抑制microRNA的功能,从而间接作用mRNA影响生物形状及蛋白表达[20]。首先使用RNAhybrid[21]预测lncRNA-microRNA相互作用对。本文综合考虑microRNA靶标结合的特征,设置RNAhybrid参数:最小自由能−25 kcal/mol,保证种子区域2~8位完全配对,p-value值小于0.05。然后,在杂交区内结合RNA结构信息[22]进行筛选:
1) microRNA序列5’端开始的10~12位必须有突起点;
2) 中间位置突起点只允许包含lncRNA序列2~4个核苷酸;
3) 除了中间位置的突起外,microRNA上的错配和G:U配对总数少于4并且连续错配小于2。
利用psRNAtarget靶向预测工具预测出microRNA-mRNA相互作用对,并且筛选出有lncRNA靶点的microRNA靶向mRNA的数据。
2.3 构建调控网络与功能预测
融合两类相互作用对,基于Cytoscape[23]工具包构建初级的lncRNA-microRNA-mRNA互作网络,然后对该网络进行模块分析,利用GO[24]数据库中的术语了解基因特性。这些GO术语被划分为3类:细胞成分(cellular component),分子功能(molecular function)和生物过程(biological process),因此可以基于GO术语对各个模块进行注释预测lncRNA的功能。
3 实验结果与分析
3.1 标注数据测试集验证结果
交叉验证(cross validation,CV)是一种模型验证技术,把给定的数据进行切分,将切分的数据集组合为训练集与测试集,用于验证模型的泛化能力,有效降低模型的过拟合问题。交叉验证方法可分为简单交叉验证法、K折交叉验证以及留一交叉验证。其中,应用最多的是K折交叉验证。标注数据即从公共数据库采集到的被验证与标注的序列数据。为了减少计算消耗的时间并评估分类模型的泛化性能,本文直接对分类模型在数据集上进行5折交叉验证,得到的性能效果作为标注数据测试集的测试结果。
为了验证本文提出方法的有效性,使用目前比较流行的CPAT、CNCI与PLEK软件在本文采用的数据集上进行分类预测,将得到的结果进行比较。CPAT使用逻辑回归模型;CNCI通过分析序列的内在组成来区分蛋白编码和非蛋白转录本,使用ATN分数矩阵以及序列结构两类特征;PLEK使用k-mer和滑动窗口来分析转录本,选取k-mers频率作为其特征。后两款软件都使用支持向量机作为其分类器,结果如表2所示。可以看出,本文提出的基于多特征融合的集成方法在精确率上超过90%,优于CPAT、CNCI与PLEK;召回率分别比CPAT、CNCI、PLEK高出6.8%、7.4%和8.8%;F1得分也优于另外三者。这些结果表明本文提出的方法可以有效地预测植物lncRNA。

表 2 基于不同方法的分类结果比较Table 2 Classification results comparison based on different methods
为了进一步验证本文基于多特征融合所构建的分类模型的有效性,分别给出单独使用开放阅读框、二级结构、k-mers作为特征训练分类器得出的预测结果,交叉验证的结果如表3所示。
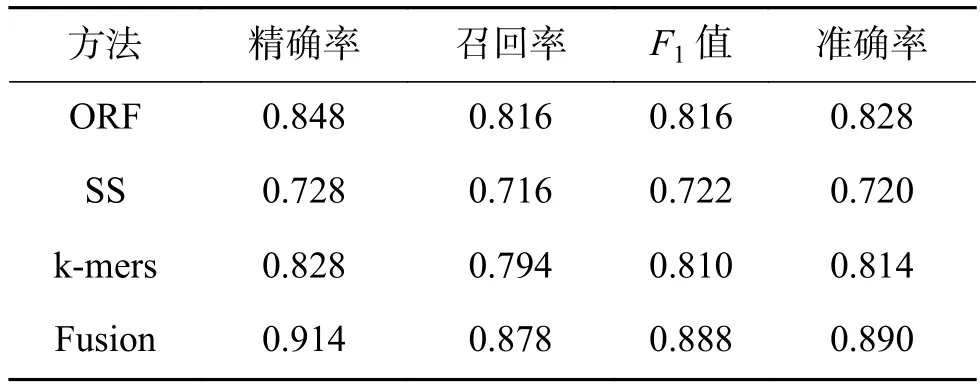
表 3 基于不同特征的分类结果比较Table 3 Classification results comparison based on different features
可以看出,本文提出的方法整体的准确率为89.0%,比单独使用开放阅读框、二级结构、kmers要分别高出6.2%、17%、7.6%,这表明提出的方法对于识别lncRNA相较于使用单一类特征是有效的。并且可以看出,使用ORF得到的预测结果要优于其他两类,这意味着ORF在识别lncRNA上具有更好的区分度。
3.2 网络构建与功能预测
经过两个靶向预测软件包的预测并且对预测结果按上述规则进行筛选后得到数据如表4~表5所示。

表 4 筛选后的microRNA-lncRNA靶点数据Table 4 Filtered microRNA-lncRNA target data

表 5 筛选后的microRNA-mRNA靶点数据Table 5 Filtered microRNA-mRNA target data
融合以上两类数据构建的初级调控网络如图1所示。
调控网络中的每个模块以microRNA为中心,形成microRNA同时与lncRNA、mRNA相互作用的调控子网络。调控子网络根据RNA作用数目和类型的不同可以分为:1)单microRNA作用网络,即单个microRNA作为结点与lncRNA、mRNA相互作用,但与网络中其他microRNA结点没有联系,如图2所示;2)多microRNA相互作用网络,不同的microRNA通过靶向同一个mRNA、lncRNA形成相互作用的模块,如图3。

图1 拟南芥初级调控网络(三角形代表microRNA,矩形代表lncRNA,圆形代表mRNA)Fig. 1 Primary regulatory network of Arabidopsis( Triangles represent microRNAs, rectangles represent lncRNAs, and circles represent mRNAs)

图2 单microRNA作用网络Fig. 2 Single microRNA interaction network
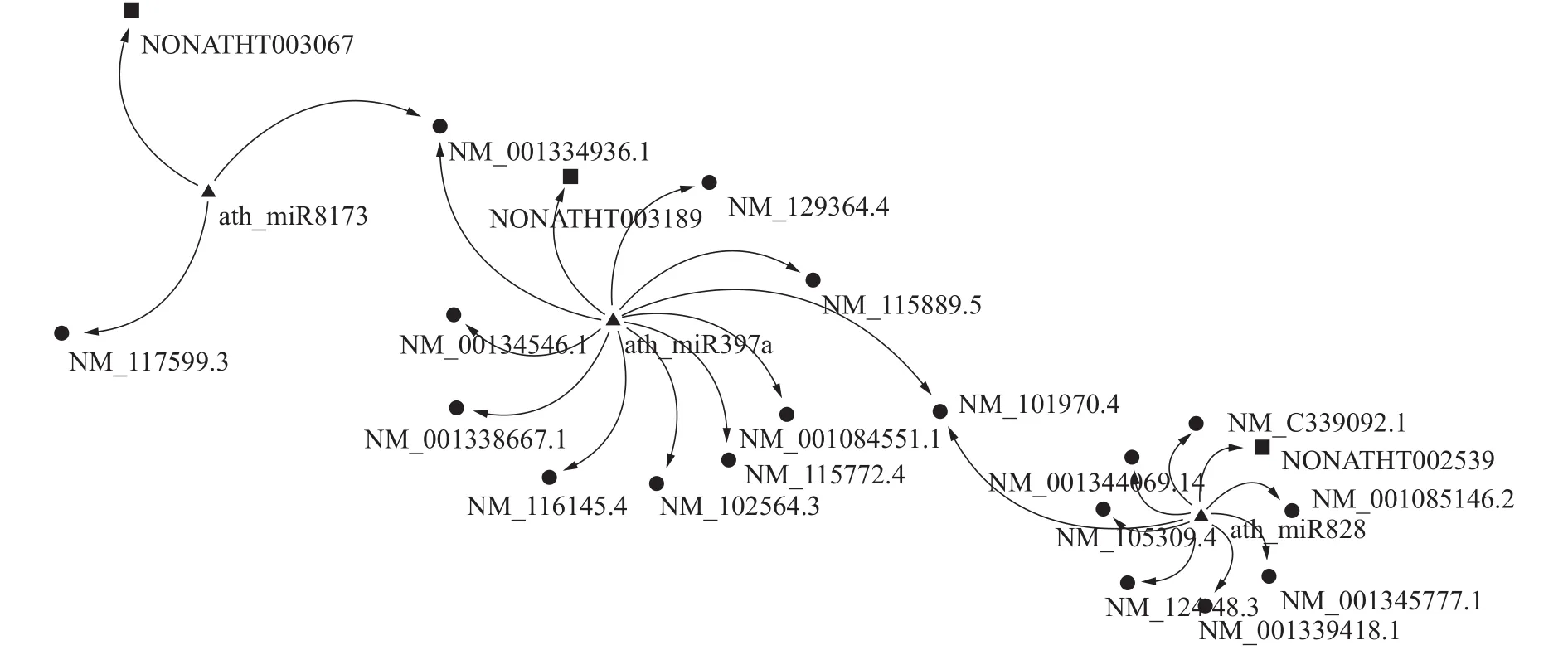
图3 多microRNA作用网络Fig. 3 Multiple microRNA interaction network
在构建调控网络并进行模块分析后,使用GO术语检查模块中的mRNA的功能注释,并对和mRNA相关的lncRNA可能参与的生物调控过程进行预测,部分结果如表6。可以看到根据相关联的RNA,本文预测的lncRNA所具有的生物调控功能。例如NONATHT002539参与到氮化合物代谢、分解代谢以及生物合成过程;NONATHT 000372促进蛋白质磷酸化;NONATHT002765和NONATHT002470、NONATHT002469都会影响细胞转化的过程等。
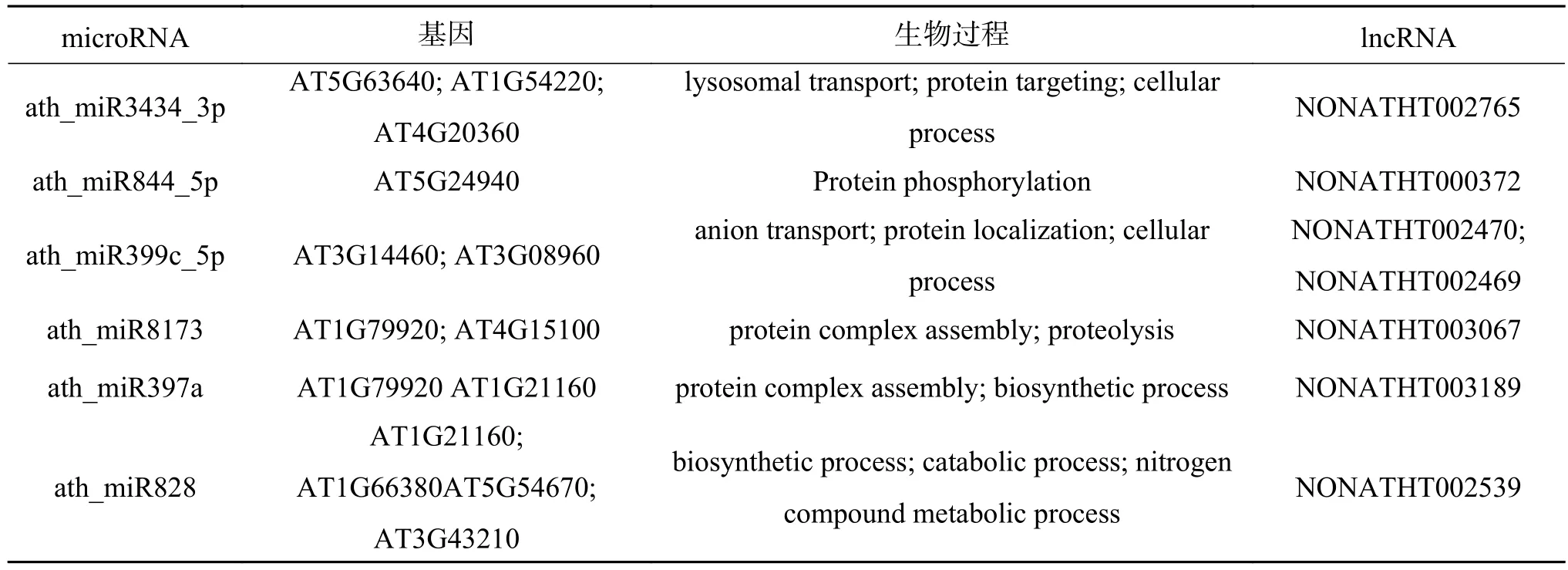
表 6 lncRNA功能预测Table 6 lncRNA function prediction
4 结束语
本文基于植物RNA序列,提取开放阅读框、二级结构和k-mers 3类特征,并将它们融合成一个90维的特征向量作为输入,训练朴素贝叶斯、支持向量机、梯度提升决策树3种机器学习模型,并采用加权投票分法来集成分类结果。通过与现有的识别软件CNCI和PLEK相比,本文提出方法取得了较好的性能,可以有效地识别预测植物lncRNA。基于内源性竞争规则,筛选lncRNA-microRNA、microRNA-mRNA相互作用数据,并整合两类数据构建调控网络,基于互作网络利用GO术语对各个模块的mRNA注释,进而通过mRNA预测lncRNA功能。未来将结合深度学习技术,进一步改善预测的准确率。
猜你喜欢
数学小灵通(1-2年级)(2021年4期)2021-06-09
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
汉字汉语研究(2020年2期)2020-08-13
电子制作(2019年22期)2020-01-14
电子技术与软件工程(2019年18期)2019-11-18
中学生数理化·七年级数学人教版(2019年4期)2019-05-20
疯狂英语·新读写(2018年3期)2018-11-29
中国惯性技术学报(2018年4期)2018-11-08
中学生数理化·七年级数学人教版(2018年6期)2018-06-26
初中生世界·七年级(2017年9期)2017-10-13
